



Oligophrenin-1 is a protein that in humans is encoded by the OPHN1 gene. [5] [6] [7]
Oligophrenin-1 is a protein that in humans is encoded by the OPHN1 gene. [5] [6] [7]
Oligophrenin 1 has 25 exons and encodes a Rho-GTPase-activating protein. The Rho proteins are important mediators of intracellular signal transduction, which affects cell migration and cell morphogenesis.
The role of OPHN1 in the medial prefrontal cortex in the behavioural responses to stress, and learned helplessness-inducing effect of OPHN1 deletion in parvalbumin interneurons, is of recent research interest. [8] [9] It is also involved in regulation in inhibitory interneurons in the olfactory bulb. [10]
Mutations in this gene are responsible for non-specific X-linked intellectual disability (previously called mental retardation). [7]
OPHN1 syndrome is a rare disorder characterized by intellectual disability and changes in the part of the brain which controls movement and balance (cerebellum). The syndrome mainly affects males. It is characterized by low muscle tone (hypotonia), developmental and cognitive delay, early-onset seizures, abnormal behavior, characteristic facial features (long face, bulging forehead, under eye creases, deep set eyes, and large ears), crossed eyes (strabismus) and inability to coordinate movements. [11] [12] A small cerebellum and large ventricles can be seen on brain imaging (MRI). [11] [13] [14] Treatment is supportive and includes physical, occupational and speech and language therapy. [15] In 2014 an OPHN1 patient organization and website was formed to support families and promote OPHN1 syndrome research. [16]
OPHN1 syndrome is caused by mutations in the OPHN1 gene, which is located on the X chromosome. Inheritance is X-linked. [11] Some females who carry a mutation in the OPHN1 gene may have mild learning disabilities, mild cognitive impairment, strabismus, and subtle facial changes. [7]

L1, also known as L1CAM, is a transmembrane protein member of the L1 protein family, encoded by the L1CAM gene. This protein, of 200-220 kDa, is a neuronal cell adhesion molecule with a strong implication in cell migration, adhesion, neurite outgrowth, myelination and neuronal differentiation. It also plays a key role in treatment-resistant cancers due to its function. It was first identified in 1984 by M. Schachner who found the protein in post-mitotic mice neurons.

FMR1 is a human gene that codes for a protein called fragile X messenger ribonucleoprotein, or FMRP. This protein, most commonly found in the brain, is essential for normal cognitive development and female reproductive function. Mutations of this gene can lead to fragile X syndrome, intellectual disability, premature ovarian failure, autism, Parkinson's disease, developmental delays and other cognitive deficits. The FMR1 premutation is associated with a wide spectrum of clinical phenotypes that affect more than two million people worldwide.

Alpha-thalassemia mental retardation syndrome (ATRX), also called alpha-thalassemia X-linked intellectual disability syndrome, nondeletion type or ATR-X syndrome, is an X-linked recessive condition associated with a mutation in the ATRX gene. Males with this condition tend to be moderately intellectually disabled and have physical characteristics including coarse facial features, microcephaly, hypertelorism, a depressed nasal bridge, a tented upper lip and an everted lower lip. Mild or moderate anemia, associated with alpha-thalassemia, is part of the condition. Females with this mutated gene have no specific signs or features, but if they do, they may demonstrate skewed X chromosome inactivation.
CDKL5 is a gene that provides instructions for making a protein called cyclin-dependent kinase-like 5 also known as serine/threonine kinase 9 (STK9) that is essential for normal brain development. Mutations in the gene can cause deficiencies in the protein. The gene regulates neuronal morphology through cytoplasmic signaling and controlling gene expression. The CDKL5 protein acts as a kinase, which is an enzyme that changes the activity of other proteins by adding a cluster of oxygen and phosphorus atoms at specific positions. Researchers are currently working to determine which proteins are targeted by the CDKL5 protein.

Rab GDP dissociation inhibitor alpha is a protein that in humans is encoded by the GDI1 gene.

Rho guanine nucleotide exchange factor 6 is a protein that, in humans, is encoded by the ARHGEF6 gene.

Lysine-specific demethylase 5C is an enzyme that in humans is encoded by the KDM5C gene. KDM5C belongs to the alpha-ketoglutarate-dependent hydroxylase superfamily.
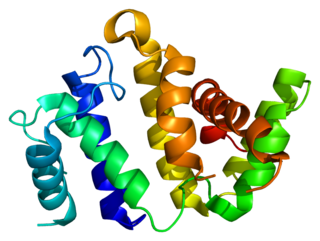
Rho GTPase activating protein 26 (ARHGAP26) also known as GTPase Regulator Associated with Focal Adhesion Kinase (GRAF) is a protein that in humans is encoded by the ARHGAP26 gene.

PHD finger protein 8 is a protein that in humans is encoded by the PHF8 gene.
X-linked intellectual disability refers to medical disorders associated with X-linked recessive inheritance that result in intellectual disability.

Gillespie syndrome, also called aniridia, cerebellar ataxia and mental deficiency, is a rare genetic disorder. The disorder is characterized by partial aniridia, ataxia, and, in most cases, intellectual disability. It is heterogeneous, inherited in either an autosomal dominant or autosomal recessive manner. Gillespie syndrome was first described by American ophthalmologist Fredrick Gillespie in 1965.

Lujan–Fryns syndrome (LFS) is an X-linked genetic disorder that causes mild to moderate intellectual disability and features described as Marfanoid habitus, referring to a group of physical characteristics similar to those found in Marfan syndrome. These features include a tall, thin stature and long, slender limbs. LFS is also associated with psychopathology and behavioral abnormalities, and it exhibits a number of malformations affecting the brain and heart. The disorder is inherited in an X-linked dominant manner, and is attributed to a missense mutation in the MED12 gene. There is currently no treatment or therapy for the underlying MED12 malfunction, and the exact cause of the disorder remains unclear.

Goldberg–Shprintzen is a very rare connective tissue condition associated with mutations in KIAA1279 gene which encodes KIF-binding protein (KBP), a protein that may interact with microtubules and actin filaments. KBP may play a key role in cytoskeleton formation and neurite growth.
Pontocerebellar hypoplasia (PCH) is a heterogeneous group of rare neurodegenerative disorders caused by genetic mutations and characterised by progressive atrophy of various parts of the brain such as the cerebellum or brainstem. Where known, these disorders are inherited in an autosomal recessive fashion. There is no known cure for PCH.

Mental retardation and microcephaly with pontine and cerebellar hypoplasia (MICPCH) – also known as mental retardation, X-linked, syndromic, Najm type (MRXSNA); X-linked intellectual deficit, Najm type; intellectual developmental disorder, X-linked, syndromic, Najm type; X-linked intellectual disability–microcephaly–pontocerebellar hypoplasia syndrome; and by variations of these terms – is a rare X-linked dominant genetic disorder of infants characterised by intellectual disability and pontocerebellar hypoplasia. It usually affects females; many males die before birth or not long after.

Jean-Louis Mandel, born in Strasbourg on February 12, 1946, is a French medical doctor and geneticist, and heads a research team at the Institute of Genetics and Molecular and Cellular Biology (IGBMC). He has been in charge of the genetic diagnosis laboratory at the University Hospitals of Strasbourg since 1992, as well as a professor at the Collège de France since 2003.

First being described and identified in 1985, Wieacker-Wolff syndrome is a rare, slowly progressive, genetic disorder present at birth and characterized by deformities of the joints of the feet, muscle degeneration, mild intellectual disability and an impaired ability to move certain muscles of the eyes, face and tongue. Wieacker syndrome is inherited as an X-linked recessive trait.
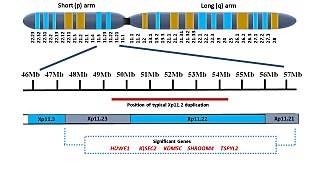
Xp11.2 duplication is a genomic variation marked by the duplication of an X chromosome region on the short arm p at position 11.2, defined by standard karyotyping (G-banding). This gene-rich, rearrangement prone region can be further divided into three loci - Xp11.21, Xp11.22 and Xp11.23. The duplication could involve any combination of these three loci. While the length of the duplication can vary from 0.5Mb to 55 Mb, most duplications measure about 4.5Mb and typically occur in the region of 11.22-11.23. Most affected females show preferential activation of the duplicated X chromosome. Features of affected individuals vary significantly, even among members of the same family. The Xp11.2 duplication can be 'silent' - presenting no obvious symptoms in carriers - which is known from the asymptomatic parents of affected children carrying the duplication. The common symptoms include intellectual disabilities, speech delay and learning difficulties, while in rare cases, children have seizures and a recognizable brain wave pattern when assessed by EEG (electroencephalography).
Christianson syndrome is an X linked syndrome associated with intellectual disability, microcephaly, seizures, ataxia and absent speech.

Stocco dos Santos syndrome is an extremely rare multi-systemic genetic disorder which is present from birth. It is characterized by heart, skeletal, muscular abnormalities with accompanying intellectual disabilities.
| | This article on a gene on the human X chromosome and/or its associated protein is a stub. You can help Wikipedia by expanding it. |