Related Research Articles
Ataxia is a neurological sign consisting of lack of voluntary coordination of muscle movements that can include gait abnormality, speech changes, and abnormalities in eye movements, that indicates dysfunction of parts of the nervous system that coordinate movement, such as the cerebellum. These nervous system dysfunctions occur in several different patterns, with different results and different possible causes. Ataxia can be limited to one side of the body, which is referred to as hemiataxia. Friedreich's ataxia has gait abnormality as the most commonly presented symptom. Dystaxia is a mild degree of ataxia.

Joubert syndrome is a rare autosomal recessive genetic disorder that affects the cerebellum, an area of the brain that controls balance and coordination.

Cerebellar hypoplasia is characterized by reduced cerebellar volume, even though cerebellar shape is (near) normal. It consists of a heterogeneous group of disorders of cerebellar maldevelopment presenting as early-onset non–progressive congenital ataxia, hypotonia and motor learning disability.
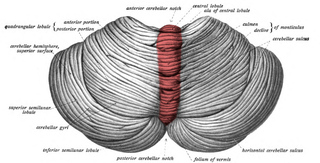
The cerebellar vermis is located in the medial, cortico-nuclear zone of the cerebellum, which is in the posterior fossa of the cranium. The primary fissure in the vermis curves ventrolaterally to the superior surface of the cerebellum, dividing it into anterior and posterior lobes. Functionally, the vermis is associated with bodily posture and locomotion. The vermis is included within the spinocerebellum and receives somatic sensory input from the head and proximal body parts via ascending spinal pathways.
Agenesis of the corpus callosum (ACC) is a rare birth defect in which there is a complete or partial absence of the corpus callosum. It occurs when the development of the corpus callosum, the band of white matter connecting the two hemispheres in the brain, in the embryo is disrupted. The result of this is that the fibers that would otherwise form the corpus callosum are instead longitudinally oriented along the ipsilateral ventricular wall and form structures called Probst bundles.

The hindbrain or rhombencephalon or lower brain is a developmental categorization of portions of the central nervous system in vertebrates. It includes the medulla, pons, and cerebellum. Together they support vital bodily processes.
Dysmetria is a lack of coordination of movement typified by the undershoot or overshoot of intended position with the hand, arm, leg, or eye. It is a type of ataxia. It can also include an inability to judge distance or scale.

Dandy–Walker malformation (DWM), also known as Dandy–Walker syndrome (DWS), is a rare congenital brain malformation in which the part joining the two hemispheres of the cerebellum does not fully form, and the fourth ventricle and space behind the cerebellum are enlarged with cerebrospinal fluid. Most of those affected develop hydrocephalus within the first year of life, which can present as increasing head size, vomiting, excessive sleepiness, irritability, downward deviation of the eyes and seizures. Other, less common symptoms are generally associated with comorbid genetic conditions and can include congenital heart defects, eye abnormalities, intellectual disability, congenital tumours, other brain defects such as agenesis of the corpus callosum, skeletal abnormalities, an occipital encephalocele or underdeveloped genitalia or kidneys. It is sometimes discovered in adolescents or adults due to mental health problems.
Cerebellar ataxia is a form of ataxia originating in the cerebellum. Non-progressive congenital ataxia (NPCA) is a classical presentation of cerebral ataxias.

22q13 deletion syndrome, also known as Phelan–McDermid syndrome (PMS), is a genetic disorder caused by deletions or rearrangements on the q terminal end of chromosome 22. Any abnormal genetic variation in the q13 region that presents with significant manifestations (phenotype) typical of a terminal deletion may be diagnosed as 22q13 deletion syndrome. There is disagreement among researchers as to the exact definition of 22q13 deletion syndrome. The Developmental Synaptopathies Consortium defines PMS as being caused by SHANK3 mutations, a definition that appears to exclude terminal deletions. The requirement to include SHANK3 in the definition is supported by many but not by those who first described 22q13 deletion syndrome.
Uner Tan syndrome (UTS) is a syndrome that was discovered by the Turkish evolutionary biologist Üner Tan. People affected by UTS walk with a quadrupedal locomotion and often have severe learning disabilities. Tan postulated that this is an example of "reverse evolution" (atavism). The proposed syndrome was featured in the 2006 BBC2 documentary The Family That Walks On All Fours.

Marinesco–Sjögren syndrome (MSS), sometimes spelled Marinescu–Sjögren syndrome, is a rare autosomal recessive disorder.

Macrocephaly-capillary malformation (M-CM) is a multiple malformation syndrome causing abnormal body and head overgrowth and cutaneous, vascular, neurologic, and limb abnormalities. Though not every patient has all features, commonly found signs include macrocephaly, congenital macrosomia, extensive cutaneous capillary malformation, body asymmetry, polydactyly or syndactyly of the hands and feet, lax joints, doughy skin, variable developmental delay and other neurologic problems such as seizures and low muscle tone.
Post-viral cerebellar ataxia also known as acute cerebellitis and acute cerebellar ataxia (ACA) is a disease characterized by the sudden onset of ataxia following a viral infection. The disease affects the function or structure of the cerebellum region in the brain.
Pontocerebellar hypoplasia (PCH) is a heterogeneous group of rare neurodegenerative disorders caused by genetic mutations and characterised by progressive atrophy of various parts of the brain such as the cerebellum or brainstem. Where known, these disorders are inherited in an autosomal recessive fashion. There is no known cure for PCH.

Autosomal recessive cerebellar ataxia type 1 (ARCA1) is a condition characterized by progressive problems with movement. Signs and symptoms of the disorder first appear in early to mid-adulthood. People with this condition initially experience impaired speech (dysarthria), problems with coordination and balance (ataxia), or both. They may also have difficulty with movements that involve judging distance or scale (dysmetria). Other features of ARCA1 include abnormal eye movements (nystagmus) and problems following the movements of objects with their eyes. The movement problems are slowly progressive, often resulting in the need for a cane, walker, or wheelchair.

Autosomal dominant cerebellar ataxia (ADCA) is a form of spinocerebellar ataxia inherited in an autosomal dominant manner. ADCA is a genetically inherited condition that causes deterioration of the nervous system leading to disorder and a decrease or loss of function to regions of the body.

Gómez–López-Hernández syndrome (GLH) or cerebellotrigeminal-dermal dysplasia is a rare neurocutaneous (Phakomatosis) disorder affecting the trigeminal nerve and causing several other neural and physical abnormalities. Gómez–López-Hernández syndrome has been diagnosed in only 34 people. Cases of Gómez–López-Hernández syndrome may be under-reported as other diseases share the characteristics of cerebellar malformation shown in Gómez–López-Hernández syndrome. Gómez–López-Hernández syndrome was first characterized in 1979.
COACH syndrome, also known as Joubert syndrome with hepatic defect, is a rare autosomal recessive genetic disease. The name is an acronym of the defining signs: cerebellar vermis aplasia, oligophrenia, congenital ataxia, coloboma and hepatic fibrosis. The condition is associated with moderate intellectual disability. It falls under the category of a Joubart Syndrome-related disorder (JSRD).
Chudley–Mccullough syndrome is a rare genetic disorder which is characterized by bilateral congenital hearing loss associated with brain malformations. It is a type of syndromic deafness.
References
- 1 2 3 4 Cotes, C; et al. (June 2015). "Congenital basis of posterior fossa anomalies". The Neuroradiology Journal. 28 (3): 238–53. doi:10.1177/1971400915576665. PMC 4757284 . PMID 26246090.
- 1 2 Toelle SP, Yalcinkaya C, Kocer N, Deonna T, Overweg-Plandsoen WC, Bast T, et al. (August 2002). "Rhombencephalosynapsis: clinical findings and neuroimaging in 9 children". Neuropediatrics. 33 (4): 209–14. doi:10.1055/s-2002-34498. PMID 12368992.
- ↑ Fernández-Jaén A, Fernández-Mayoralas DM, Calleja-Pérez B, Muñoz-Jareño N, Moreno N (January 2009). "Gomez-Lopez-Hernandez syndrome: two new cases and review of the literature". Pediatric Neurology. 40 (1): 58–62. doi:10.1016/j.pediatrneurol.2008.10.001. PMID 19068257.
- 1 2 3 Kotetishvili B, Makashvili M, Okujava M, Kotetishvili A, Kopadze T (August 2018). "Co-occurrence of Gomez-Lopez-Hernandez syndrome and Autism Spectrum Disorder: Case report with review of literature". Intractable & Rare Diseases Research. 7 (3): 191–195. doi:10.5582/irdr.2018.01062. PMC 6119670 . PMID 30181940.
- ↑ Mak, CCY; et al. (13 December 2019). "MN1 C-terminal truncation syndrome is a novel neurodevelopmental and craniofacial disorder with partial rhombencephalosynapsis". Brain: A Journal of Neurology. 143 (1): 55–68. doi:10.1093/brain/awz379. PMC 7962909 . PMID 31834374.
- ↑ Aldinger, KA; et al. (December 2018). "Rhombencephalosynapsis: Fused cerebellum, confused geneticists". American Journal of Medical Genetics. Part C, Seminars in Medical Genetics. 178 (4): 432–439. doi:10.1002/ajmg.c.31666. PMC 6540982 . PMID 30580482.
- 1 2 Tan TY, McGillivray G, Goergen SK, White SM (November 2005). "Prenatal magnetic resonance imaging in Gomez-Lopez-Hernandez syndrome and review of the literature". American Journal of Medical Genetics. Part A. 138 (4): 369–73. doi:10.1002/ajmg.a.30967. PMID 16158443. S2CID 11532423.
- ↑ Truwit, CL; et al. (1991). "MR imaging of rhombencephalosynapsis: report of three cases and review of the literature". AJNR. American Journal of Neuroradiology. 12 (5): 957–65. PMC 8333516 . PMID 1950929.