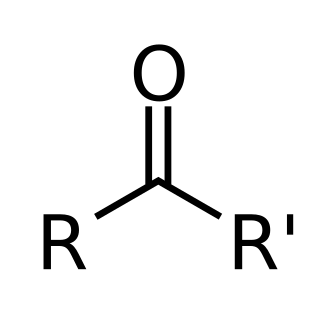
In organic chemistry, a ketone is an organic compound with the structure R−C(=O)−R', where R and R' can be a variety of carbon-containing substituents. Ketones contain a carbonyl group −C(=O)−. The simplest ketone is acetone, with the formula (CH3)2CO. Many ketones are of great importance in biology and in industry. Examples include many sugars (ketoses), many steroids, and the solvent acetone.

The aldol reaction is a reaction in organic chemistry that combines two carbonyl compounds to form a new β-hydroxy carbonyl compound. Its simplest form might involve the nucleophilic addition of an enolized ketone to another:

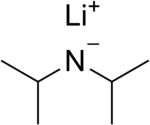
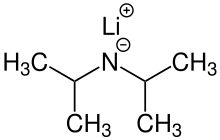
In organometallic chemistry, organolithium reagents are chemical compounds that contain carbon–lithium (C–Li) bonds. These reagents are important in organic synthesis, and are frequently used to transfer the organic group or the lithium atom to the substrates in synthetic steps, through nucleophilic addition or simple deprotonation. Organolithium reagents are used in industry as an initiator for anionic polymerization, which leads to the production of various elastomers. They have also been applied in asymmetric synthesis in the pharmaceutical industry. Due to the large difference in electronegativity between the carbon atom and the lithium atom, the C−Li bond is highly ionic. Owing to the polar nature of the C−Li bond, organolithium reagents are good nucleophiles and strong bases. For laboratory organic synthesis, many organolithium reagents are commercially available in solution form. These reagents are highly reactive, and are sometimes pyrophoric.

In organic chemistry, alkenols are a type of reactive structure or intermediate in organic chemistry that is represented as an alkene (olefin) with a hydroxyl group attached to one end of the alkene double bond. The terms enol and alkenol are portmanteaus deriving from "-ene"/"alkene" and the "-ol" suffix indicating the hydroxyl group of alcohols, dropping the terminal "-e" of the first term. Generation of enols often involves deprotonation at the α position to the carbonyl group—i.e., removal of the hydrogen atom there as a proton H+. When this proton is not returned at the end of the stepwise process, the result is an anion termed an enolate. The enolate structures shown are schematic; a more modern representation considers the molecular orbitals that are formed and occupied by electrons in the enolate. Similarly, generation of the enol often is accompanied by "trapping" or masking of the hydroxy group as an ether, such as a silyl enol ether.
The Robinson annulation is a chemical reaction used in organic chemistry for ring formation. It was discovered by Robert Robinson in 1935 as a method to create a six membered ring by forming three new carbon–carbon bonds. The method uses a ketone and a methyl vinyl ketone to form an α,β-unsaturated ketone in a cyclohexane ring by a Michael addition followed by an aldol condensation. This procedure is one of the key methods to form fused ring systems.
In organic chemistry, enolates are organic anions derived from the deprotonation of carbonyl compounds. Rarely isolated, they are widely used as reagents in the synthesis of organic compounds.
In organic chemistry, self-condensation is an organic reaction in which a chemical compound containing a carbonyl group acts both as the electrophile and the nucleophile in an aldol condensation. It is also called a symmetrical aldol condensation as opposed to a mixed aldol condensation in which the electrophile and nucleophile are different species.
As the name suggests, a non-nucleophilic base is a sterically hindered organic base that is a poor nucleophile. Normal bases are also nucleophiles, but often chemists seek the proton-removing ability of a base without any other functions. Typical non-nucleophilic bases are bulky, such that protons can attach to the basic center but alkylation and complexation is inhibited.
The Claisen condensation is a carbon–carbon bond forming reaction that occurs between two esters or one ester and another carbonyl compound in the presence of a strong base. The reaction produces a β-keto ester or a β-diketone. It is named after Rainer Ludwig Claisen, who first published his work on the reaction in 1887. The reaction has often been displaced by diketene-based chemistry, which affords acetoacetic esters.

n-Butyllithium C4H9Li (abbreviated n-BuLi) is an organolithium reagent. It is widely used as a polymerization initiator in the production of elastomers such as polybutadiene or styrene-butadiene-styrene (SBS). Also, it is broadly employed as a strong base (superbase) in the synthesis of organic compounds as in the pharmaceutical industry.
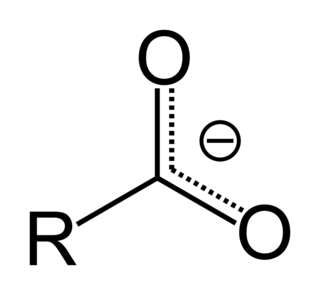
In organic chemistry, a carboxylate is the conjugate base of a carboxylic acid, RCOO−. It is an ion with negative charge.
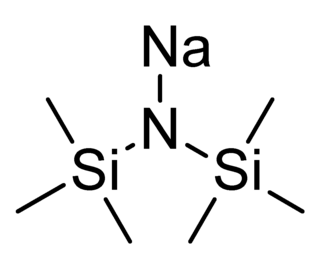
Sodium bis(trimethylsilyl)amide is the organosilicon compound with the formula NaN(Si 3)2. This species, usually called NaHMDS, is a strong base used for deprotonation reactions or base-catalyzed reactions. Its advantages are that it is commercially available as a solid and it is soluble not only in ethers, such as THF or diethyl ether, but also in aromatic solvents, like benzene and toluene by virtue of the lipophilic TMS groups.

In stereochemistry, a chiral auxiliary is a stereogenic group or unit that is temporarily incorporated into an organic compound in order to control the stereochemical outcome of the synthesis. The chirality present in the auxiliary can bias the stereoselectivity of one or more subsequent reactions. The auxiliary can then be typically recovered for future use.
The E1cB elimination reaction is a type of elimination reaction which occurs under basic conditions, where the hydrogen to be removed is relatively acidic, while the leaving group is a relatively poor one. Usually a moderate to strong base is present. E1cB is a two-step process, the first step of which may or may not be reversible. First, a base abstracts the relatively acidic proton to generate a stabilized anion. The lone pair of electrons on the anion then moves to the neighboring atom, thus expelling the leaving group and forming a double or triple bond. The name of the mechanism - E1cB - stands for Elimination Unimolecular conjugate Base. Elimination refers to the fact that the mechanism is an elimination reaction and will lose two substituents. Unimolecular refers to the fact that the rate-determining step of this reaction only involves one molecular entity. Finally, conjugate base refers to the formation of the carbanion intermediate, which is the conjugate base of the starting material.

tert-Butyllithium is a chemical compound with the formula (CH3)3CLi. As an organolithium compound, it has applications in organic synthesis since it is a strong base, capable of deprotonating many carbon molecules, including benzene. tert-Butyllithium is available commercially as solutions in hydrocarbons (such as pentane); it is not usually prepared in the laboratory.

Benzylideneacetone is the organic compound described by the formula C6H5CH=CHC(O)CH3. Although both cis- and trans-isomers are possible for the α,β-unsaturated ketone, only the trans isomer is observed. Its original preparation demonstrated the scope of condensation reactions to construct new, complex organic compounds. Benzylideneacetone is used as a flavouring ingredient in food and perfumes.
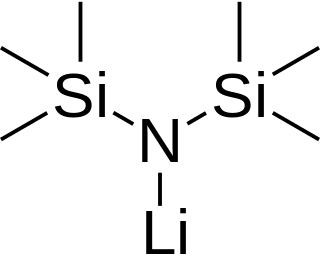
Lithium bis(trimethylsilyl)amide is a lithiated organosilicon compound with the formula LiN(Si(CH3)3)2. It is commonly abbreviated as LiHMDS or Li(HMDS) (lithium hexamethyldisilazide - a reference to its conjugate acid HMDS) and is primarily used as a strong non-nucleophilic base and as a ligand. Like many lithium reagents, it has a tendency to aggregate and will form a cyclic trimer in the absence of coordinating species.
The Chichibabin reaction is a method for producing 2-aminopyridine derivatives by the reaction of pyridine with sodium amide. It was reported by Aleksei Chichibabin in 1914. The following is the overall form of the general reaction:
Heteroatom-promoted lateral lithiation is the site-selective replacement of a benzylic hydrogen atom for lithium for the purpose of further functionalization. Heteroatom-containing substituents may direct metalation to the benzylic site closest to the heteroatom or increase the acidity of the ring carbons via an inductive effect.

Alpha-substitution reactions occur at the position next to the carbonyl group, the α-position, and involve the substitution of an α hydrogen atom by an electrophile, E, through either an enol or enolate ion intermediate.