
Mitochondrial trifunctional protein deficiency is an autosomal recessive fatty acid oxidation disorder that prevents the body from converting certain fats to energy, particularly during periods without food. People with this disorder have inadequate levels of an enzyme that breaks down a certain group of fats called long-chain fatty acids.

Very long-chain specific acyl-CoA dehydrogenase, mitochondrial (VLCAD) is an enzyme that in humans is encoded by the ACADVL gene.

ACADM is a gene that provides instructions for making an enzyme called acyl-coenzyme A dehydrogenase that is important for breaking down (degrading) a certain group of fats called medium-chain fatty acids.

Short-chain acyl-coenzyme A dehydrogenase deficiency (SCADD) is an autosomal recessive fatty acid oxidation disorder which affects enzymes required to break down a certain group of fats called short chain fatty acids.
Acyl-CoA dehydrogenases (ACADs) are a class of enzymes that function to catalyze the initial step in each cycle of fatty acid β-oxidation in the mitochondria of cells. Their action results in the introduction of a trans double-bond between C2 (α) and C3 (β) of the acyl-CoA thioester substrate. Flavin adenine dinucleotide (FAD) is a required co-factor in addition to the presence of an active site glutamate in order for the enzyme to function.

Acyl-CoA dehydrogenase, C-2 to C-3 short chain is an enzyme that in humans is encoded by the ACADS gene. This gene encodes a tetrameric mitochondrial flavoprotein, which is a member of the acyl-CoA dehydrogenase family. This enzyme catalyzes the initial step of the mitochondrial fatty acid beta-oxidation pathway. The ACADS gene associated with short-chain acyl-coenzyme A dehydrogenase deficiency.

Acyl-CoA dehydrogenase, long chain is a protein that in humans is encoded by the ACADL gene.
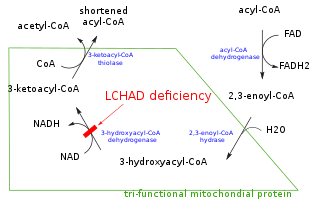
Mitochondrial trifunctional protein (MTP) is a protein attached to the inner mitochondrial membrane which catalyzes three out of the four steps in beta oxidation. MTP is a hetero-octamer composed of four alpha and four beta subunits:

Trifunctional enzyme subunit beta, mitochondrial (TP-beta) also known as 3-ketoacyl-CoA thiolase, acetyl-CoA acyltransferase, or beta-ketothiolase is an enzyme that in humans is encoded by the HADHB gene.

ACADSB is a human gene that encodes short/branched chain specific acyl-CoA dehydrogenase (SBCAD), an enzyme in the acyl CoA dehydrogenase family.

Electron-transferring-flavoprotein dehydrogenase is an enzyme that transfers electrons from electron-transferring flavoprotein in the mitochondrial matrix, to the ubiquinone pool in the inner mitochondrial membrane. It is part of the electron transport chain. The enzyme is found in both prokaryotes and eukaryotes and contains a flavin and FE-S cluster. In humans, it is encoded by the ETFDH gene. Deficiency in ETF dehydrogenase causes the human genetic disease multiple acyl-CoA dehydrogenase deficiency.

Pyruvate dehydrogenase E1 component subunit alpha, somatic form, mitochondrial is an enzyme that in humans is encoded by the PDHA1 gene.The pyruvate dehydrogenase complex is a nuclear-encoded mitochondrial matrix multienzyme complex that provides the primary link between glycolysis and the tricarboxylic acid (TCA) cycle by catalyzing the irreversible conversion of pyruvate into acetyl-CoA. The PDH complex is composed of multiple copies of 3 enzymes: E1 (PDHA1); dihydrolipoyl transacetylase (DLAT) ; and dihydrolipoyl dehydrogenase (DLD). The E1 enzyme is a heterotetramer of 2 alpha and 2 beta subunits. The E1-alpha subunit contains the E1 active site and plays a key role in the function of the PDH complex.

NADH dehydrogenase [ubiquinone] iron-sulfur protein 4, mitochondrial (NDUFS4) also known as NADH-ubiquinone oxidoreductase 18 kDa subunit is an enzyme that in humans is encoded by the NDUFS4 gene. This gene encodes a nuclear-encoded accessory subunit of the mitochondrial membrane respiratory chain NADH dehydrogenase. Complex I removes electrons from NADH and passes them to the electron acceptor ubiquinone. Mutations in this gene can cause mitochondrial complex I deficiencies such as Leigh syndrome.

The human ETFA gene encodes the Electron-transfer-flavoprotein, alpha subunit, also known as ETF-α. Together with Electron-transfer-flavoprotein, beta subunit, encoded by the 'ETFB' gene, it forms the heterodimeric electron transfer flavoprotein (ETF). The native ETF protein contains one molecule of FAD and one molecule of AMP, respectively.

The human ETFB gene encodes the Electron-transfer-flavoprotein, beta subunit, also known as ETF-β. Together with Electron-transfer-flavoprotein, alpha subunit, encoded by the 'ETFA' gene, it forms the heterodimeric Electron transfer flavoprotein (ETF). The native ETF protein contains one molecule of FAD and one molecule of AMP, respectively.
NADH-ubiquinone oxidoreductase 75 kDa subunit, mitochondrial (NDUFS1) is an enzyme that in humans is encoded by the NDUFS1 gene. The encoded protein, NDUFS1, is the largest subunit of complex I, located on the inner mitochondrial membrane, and is important for mitochondrial oxidative phosphorylation. Mutations in this gene are associated with complex I deficiency.

(See also: List of proteins in the human body)

Complex I intermediate-associated protein 30, mitochondrial (CIA30), or NADH dehydrogenase [ubiquinone] 1 alpha subcomplex assembly factor 1 (NDUFAF1), is a protein that in humans is encoded by the NDUFAF1 or CIA30 gene. The NDUFAF1 gene encodes a human homolog of a Neurospora crassa protein involved in the assembly of complex I. The NDUFAF1 protein is an assembly factor of NADH dehydrogenase (ubiquinone) also known as complex I, which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain. Variants of the NDUFAF1 gene are associated with hypertrophic cardiomyopathy, leukodystrophy, and cardioencephalomyopathy.

NADH dehydrogenase [ubiquinone] 1 alpha subcomplex assembly factor 3, also known as 2P1, E3-3, or C3orf60, is a protein that in humans is encoded by the NDUFAF3 gene. NDUFAF3 is a mitochondrial assembly protein involved in the assembly of NADH dehydrogenase (ubiquinone) also known as complex I, which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain. Mutations in this gene have been associated with severe complex I deficiency and Leigh syndrome.
NADH:ubiquinone oxidoreductase complex assembly factor 2 (NDUFAF2), also known as B17.2L or NDUFA12L is a protein that in humans is encoded by the NDUFAF2, or B17.2L, gene. The NDUFAF2 protein is a chaperone involved in the assembly of NADH dehydrogenase (ubiquinone) also known as complex I, which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain. Mutations in this gene have been associated with progressive encephalopathy and Leigh disease resulting from mitochondrial complex I deficiency.



