Methods for the estimation of the abundance of bacterioplankton in a specific body of water
Bacterioplankton counting is the estimation of the abundance of bacterioplankton in a specific body of water, which is useful information to marine microbiologists. Various counting methodologies have been developed over the years to determine the number present in the water being observed. Methods used for counting bacterioplankton include epifluorescence microscopy, flow cytometry, measures of productivity through frequency of dividing cells (FDC), thymidine incorporation, and leucine incorporation.
Factors such as salinity, temperature, latitude, various nutrient levels, water movement and the presence of other organisms can affect bacterioplankton enumeration.[1][2][3][4][5] Changes in these factors affect the bacterioplankton count, causing it to vary by body of water, location, distance from shore and season.[6][7][8]
Bacterioplankton count is usually expressed in cells per ml (cells ml−1).
Uses
In understanding marine microbiology and the aquatic ecosystem, bacterioplankton counts can be useful. Observation of bacterioplankton number can provide more information in the following:
Processes involved in various nutrient cycling in the aquatic systems[9][10]
For determining environmental changes, especially extremes [12]
Variation in bacterioplankton count not derived by seasonal adjustments can provide correlation with environmental stresses such as having a significant shift in nutrient levels within a body of water[13][14][15]
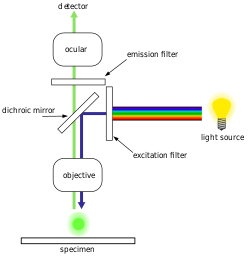
Epifluorescence microscopy is an advanced optical microscope technique that relies on the use of fluorescent dyes that bind to specific biological markers, which then emit a distinctive emission spectra that is identified through the lens. Fluorescent dyes include DAPI, Acridine Orange, SYBR Green 1, and YO-PRO-1, all of which are capable of staining both DNA and RNA structures in biological samples such as bacteria and viruses.[18][19][20][21] However, DNA staining is primarily used for bacterial cell identification. With modern epifluorescence microscopy, the industry standard for estimating and counting bacterial cell quantities is by the use of a DAPI stain.[22] This technique can be performed for samples from a wide range of environments and locations, such as seawater, various sources of freshwater, as well as soils and sediments.[22]
Enumeration technique
In a standard experiment, prepared bacterial samples are placed onto counting slides and then viewed under an epifluorescence microscope. Magnification is set to a level where the 0.1 X 0.1mm square units on the counting slide are clearly visible.[23] To quantify the bacteria, cells are counted in 5-30 random square unit field-of-views and an average bacteria count per field is tabulated.[22] This value is then extrapolated to estimate the total bacterial cell-count per mL by determining the total number of fields-of-view on the slide deposition area and multiplying this by the average bacterial count per counting unit.[23]
Reliability
To enumerate bacterial cell quantities, only small portions of bacteria in a sample are physically counted for logistical reasons, upon which total abundances are estimated by extrapolation. Mean values are then used for comparison among samples. However the accuracy of this technique, where tabulation of only a small subset is used to estimate total abundance quantities, has been brought into question.[22] Primarily, it has been shown that the distribution of bacterial cells on counting slides can be uneven and inconsistent.[22] In addition, to get a legitimate estimate of bacterial counts by using this technique, it has been suggested that more than 350 individual cells, from 20 fields of view must be measured.[22] This can be not only time-consuming, but difficult to achieve in certain samples.
Flow cytometric analysis (or, flow cytometry) is a common procedure in many clinical applications. However, despite its discovery more than three decades ago, its adoption by aquatic microbial ecology in enumeration of bacterioplankton, has been relatively slow.[24] Its use is yet to surpass epifluorescence microscopy.[25] Despite both abundance estimation techniques being relatively accurate, flow cytometry is less prone to human error, more precise, pertains a higher resolution and is capable of examining tens of thousands of cells in a matter of minutes.[24] Flow cytometry is also able to provide information regarding size, activity and morphology of cells besides abundance of cells.[26]
Flow cytometry can be used to distinguish and quantify both photosynthetic and non-photosynthetic bacterioplankton.[26] Quantification of photosynthetic prokaryotes such as cyanobacteria and picoeukaryotic algae is made possible by the ability of photosynthetic pigments to fluoresce.[27] For instance, the different formation of photosynthetic pigments in the two major photosynthetic prokaryotes, Prochlorococcus and Synechococcus, enable their very distinction.[28][29][30]Prochlorococcus contains divynyl-chlorophylls a and b which display solely red fluorescence under excitation by blue or UV light, while Synechococcus emits both orange and red fluorescence; orange from phycobilins and red from chlorophyll. Besides fluorescence, Prochlorococcus and Synechococus are of significantly different sizes and hence deliver different scatter signals upon flow cytometric analysis. This further helps in their differentiation.[31] Quantification of prochlorococcus is considered a major breakthrough as it has almost only been possible through flow cytometry. This is due to the inability of epifluorescence microscopy to detect the low chlorophyll autofluorescence present in Prochlorococcus.[26]
Besides photosynthetic bacterioplankton, non-photosynthetic bacterioplankton can also be enumerated by flow cytometry. This is done via DNA or food vacuole staining.[27] Flow cytometry has especially been successful at differentiating Prochloroccocus from heterotrophic bacteria, whose counts were initially confounded due to their similar size.
Use of epifluorescence microscopy over flow cytometry in many microbial ecology labs can be blamed on a number of economic and practical factors. First, the use of commercial flow cytometers requires the expertise of a rigorously trained technician. Second, flow cytometers are fairly expensive in comparison to epi-fluorescence microscopy apparatus. Third, many flow cytometers are designed to examine blood cells; oceanic bacteria are relatively small and hence approach limit of resolution in many commercial flow cytometers.[32]
Enumeration Process
Flow cytometric quantification of bacterioplankton involves four steps: fixation, staining, data processing and data interpretation.
Fixation
Fixation is done to not only preserve sample, but also to increase permeability of cells to stains.[24] However, most common fixation agents have the capacity to alter cells by changing certain aspects such as size, how light is scattered, autofluorescence and nucleic acids. This is problematic as flow cytometric distinction of cells relies on these qualities. Some fixatives also lead to complete loss of cells.[24] Presently, some of the agents used in the fixation process include two variations of formaldehyde (formalin and paraformaldehyde), 70% ethanol, glutaraldehyde and TCA.[33] It is presumed that the best fixation agent for protein and nucleic acids is paraformaldehyde due to its ability to swiftly enter cells.[24]
Staining
In flow cytometry, staining enables distinction of bacterioplankton from non-bacterial particles. It involves the incubation of sample in the wide array of fluorochromes such as UV-excited dyes (DAPI and Hoechst 33342) and blue-light-excited nucleic acid dyes (TO-PRO-1,TOTO-1, SYBR Green I).[31] For a long time, flow cytometers utilized UV-excited dyes to examine bacterioplankton which could be used in either low-cost flow cytometers with limited sensitivity, or expensive flow cytometers with the high sensitivity needed to distinguish heterotrophic bacteria from autotrophs. The introduction of blue-excited dyes such as SYBR Green I, enabled high quality flow cytometric analysis of bacterioplankton on low-cost, high-sensitivity flow cytometers.[31]
Incubation times for optimum staining varies from compound to compound. UV-excited dyes can require an hour or more while blue-light-excited dyes require a mere 15 minutes.[24]
Staining can be accompanied by buffers such as Triton X-100 which make cells more permeable to stains. They are especially used in cell-impermeant dyes like TO-PRO-1. Buffers are also used to dilute dyes sensitive to ionic strength such as Picogreen, YO-PRO-1 and YOYO-1. The use of buffers however, can be harmful to cells as buffers like Triton-X-100 can not only extinguish chlorophyll fluorescence, but also create unwanted background fluorescence. This can increase the difficulty of distinguishing between heterotrophic bacteria and autotrophic prokaryotes.[24]
In a flow cytometric analysis, over 200 cells pass in front of a laser beam or mercury lamp every second, a cell at a time. Photomultipliers gather the amount of light each particle scatters and the fluorescence emitted upon excitation. This information is then internalized and interpreted by the system as an event. However, despite the ability of flow cytometers to count cells with very little effort, most have no way of determining actual concentration of cells. This can be determined through a variety of methods including, use of reference beads whose quantity is pre-determined (helps determine the ratio of bacteria to beads), weight measurements before and after experiment and daily calibration of flow.[24]
A big advantage of flow cytometers is their ability to identify different populations of bacterioplankton. This discrimination is done via analysis of four factors; light scatter, green fluorescence, blue fluorescence and red fluorescence. Light scatter analysis is inadequate alone and is often examined alongside fluorescence for a number of reasons; first, sea water contains many particles that scatter light like bacteria. Second, the sizes of many oceanic bacteria approach limit of resolution. The amount of light scattered by cells is determined by not only size of cells, but also internal structure, refractive index, shape and orientation of particle. Scattered light is classified into either forward scatter (FSC) or side scatter (SSC). The former has been associated with cell volume and mass while the latter has been associated with index of refraction, content and granularity of cells [24]
When cell concentrations are higher than 2.5 million cells/mL, the likelihood of more than once cell passing in close proximity and being recorded as a single event is magnified. This is known as coincidence and can be easily avoided by diluting sample before hand[31]
Measures of productivity
Frequency of dividing cells
Frequency of dividing cells (FDC) is a method used to predict the average growth rate of an aquatic heterotrophic bacterial community.[34] The method uses cell division, specifically septum formation, as a proxy for growth rate.[34] Cells are considered divided, when cavities between individual cells (invagination) are observed under epifluorescence microscopy.[34] FDC is based on the assumption that there relationship between the proportion of cells currently dividing and the growth rate in a bacterial community.[35]
Thymidine incorporation
Thymidine incorporation is one of the most extensively used methods to estimate bacterial growth.[36] Thymidine is a precursor for DNA, and DNA synthesis can be measured by tritiated thymidine incorporation into nucleic acids.[37] Thymidine incorporation measures growth based on rates of DNA synthesis, using the assumption that only growing cells can incorporate the radioactive thymidine to synthesize DNA.[38]
Weaknesses of this procedure include labeling of other molecules besides DNA when tritiated thymidine is added to a sample.[36] In cases of carbon limitation, thymidine may also be used as a carbon source instead of as a DNA precursor.[36] Results of thymidine incorporation experiments may be misleading when the proportion of thymidine incorporated into DNA compared to other molecules is not known.[36]
Leucine incorporation
Leucine incorporation is used as a measure of protein synthesis in aquatic bacteria communities.[39] Radio-labeled leucine is added to samples, and its accumulation into proteins, the hot trichloroacetic acid (CA)-insoluble parts of the cell is determined.[39] The samples are then collected on membrane filter.[39] Leucine protein is taken up by more than 50% of aquatic bacterial populations, and leucine incorporation can be used to estimate nitrogen utilization in the bacterial community.[39]
Marine seasonal succession dynamics
As bacterial populations have unique metabolisms and resource preferences, the use of high-resolution time-series analysis of bacterial compositions allows for the identification of patterns in seasonal bacterial succession.[40] Differences in bacterial community compositions give rise to particular permutations of interspecies bacterial interactions with photosynthetic plankton, protist grazers, and phages thereby impacting seasonality dynamics. Statistical methods used to verify patterns in population dynamics and composition are demonstrated to be replicable over some years, and environmental factors served as predictors of these temporal patterns.[41]
Seasonal succession in temperate regions
As seasonal successions of phytoplankton populations follow a consistent recurring pattern, bacterial dynamics and phytoplankton succession can be correlated.[40] In general, seasonal changes in bacterial composition follow changes in temperature and chlorophyll a, while nutrient availability limits bacterioplankton growth rates.[42][43][44][45][6][46] During water column mixing in late autumn/winter, nutrients brought to the surface kicks start a distinct diatom spring bloom followed by dinoflagellates.[40] After the spring bloom, bacterial production and growth become elevated due to the release of Dissolved organic matter (DOM) from phytoplankton decay.[47][48] In this early succession stage, members of the class Flavobacteria (Bacteroidetes) are typically the dominant components of the bacterial community.[49][50] Genome analysis and meta-transcriptomics have uncovered the presence of bacteria containing multiple hydrolytic enzymes facilitating the degradation and assimilation of DOM.[51][52][53][54] During spring blooms, some members of the Roseobacter clade (Alphaproteobacteria) and some Gammaproteobacteria are usually associated with DOM degradation.[48][49] As temperatures increase and the nutrients from the spring bloom gets depleted, smaller phytoplankton and cyanobacteria grow in the now oligotrophic waters.[40]
As waters become stratified in summer, Roseobacter, SAR86 (Gammaproteobacteria), and SAR11 (Alphaproteobacteria) clades of bacteria increase in abundance.[55][56] The frequently observed autumn diatom and dinoflagellate blooms are correlated with supplementary nutrient inputs and high-frequency sampling in the Baltic Sea found that in autumn, Actinomycetota generally increase followed by different autumn-specific Flavobacteria, SAR11, and Planctomycetota.[49]
In the Mediterranean Sea, deep winter mixing allows members of the SAR11 clade to achieve increased diversity as the oligotrophic populations that once dominated during the summer stratification die off slowly.[57] Among archaea in the Mediterranean Sea, Nitrososphaerota (formerly Thaumarchaeota) Marine Group I (MGI) and Euryarchaeota Marine Group II (MGII.B) populations became dominant in winter.[58] While in the Baltic Sea, winter mixing brings Campylobacterota and archaea populations to the surface from their deep habitat.[49]
↑ He J, Zhang F, Lin L, Ma Y, Chen J (2012). "Bacterioplankton and picophytoplankton abundance, biomass, and distribution in the Western Canada Basin during summer 2008". Deep Sea Research Part II: Topical Studies in Oceanography. 81–84: 36–45. Bibcode:2012DSRII..81...36H. doi:10.1016/j.dsr2.2012.08.018.
↑ López-Flores R, Boix D, Badosa A, Brucet S, Quintana XD (2009). "Environmental factors affecting bacterioplankton and phytoplankton dynamics in confined Mediterranean salt marshes (NE Spain)". Journal of Experimental Marine Biology and Ecology. 369 (2): 118–126. Bibcode:2009JEMBE.369..118L. doi:10.1016/j.jembe.2008.11.003.
↑ Medvinsky, Alexander B.; Adamovich, Boris V.; Aliev, Rubin R.; Chakraborty, Amit; Lukyanova, Elena V.; Mikheyeva, Tamara M.; Nikitina, Ljudmila V.; Nurieva, Nailya I.; Rusakov, Alexey V. (2017). "Temperature as a factor affecting fluctuations and predictability of the abundance of lake bacterioplankton". Ecological Complexity. 32: 90–98. Bibcode:2017EcoCm..32...90M. doi:10.1016/j.ecocom.2017.10.002.
↑ Currie DJ, Kalff J (March 1984). "The relative importance of bacterioplankton and phytoplankton in phosphorus uptake in freshwater1". Limnology and Oceanography. 29 (2): 311–321. Bibcode:1984LimOc..29..311C. doi:10.4319/lo.1984.29.2.0311.
↑ Zhang D, Wang X, Xiong J, Zhu J, Wang Y, Zhao Q, Chen H, Guo A, Wu J (2014). "Bacterioplankton assemblages as biological indicators of shrimp health status". Ecological Indicators. 38: 218–224. Bibcode:2014EcInd..38..218Z. doi:10.1016/j.ecolind.2013.11.002.
↑ Tanious FA, Veal JM, Buczak H, Ratmeyer LS, Wilson WD (1992-03-31). "DAPI (4',6-diamidino-2-phenylindole) binds differently to DNA and RNA: minor-groove binding at AT sites and intercalation at AU sites". Biochemistry. 31 (12): 3103–3112. doi:10.1021/bi00127a010. PMID1372825.
↑ Gonzalez K, Mcvey S, Cunnick J, Udovichenko IP, Takemoto DJ (1995). "Acridine orange differential staining of total DNA and RNA in normal and galactosemic lens epithelial cells in culture using flow cytometry". Current Eye Research. 14 (4): 269–273. doi:10.3109/02713689509033525. PMID7541739.
1 2 3 Sieracki, Michael E.; Haugen, Elin M.; Cucci, Terry L. (1995-08-01). "Overestimation of heterotrophic bacteria in the Sargasso Sea: direct evidence by flow and imaging cytometry". Deep Sea Research Part I: Oceanographic Research Papers. 42 (8): 1399–1409. Bibcode:1995DSRI...42.1399S. doi:10.1016/0967-0637(95)00055-B. ISSN0967-0637.
↑ Chisholm, Sallie W.; Frankel, Sheila L.; Goericke, Ralf; Olson, Robert J.; Palenik, Brian; Waterbury, John B.; West-Johnsrud, Lisa; Zettler, Erik R. (1992-02-01). "Prochlorococcus marinus nov. gen. nov. sp.: an oxyphototrophic marine prokaryote containing divinyl chlorophyll a and b". Archives of Microbiology. 157 (3): 297–300. Bibcode:1992ArMic.157..297C. doi:10.1007/BF00245165. ISSN0302-8933. S2CID32682912.
↑ Chisholm, Sallie W.; Olson, Robert J.; Zettler, Erik R.; Goericke, Ralf; Waterbury, John B.; Welschmeyer, Nicholas A. (July 1988). "A novel free-living prochlorophyte abundant in the oceanic euphotic zone". Nature. 334 (6180): 340–343. Bibcode:1988Natur.334..340C. doi:10.1038/334340a0. ISSN1476-4687. S2CID4373102.
1 2 Buchan A, LeCleir GR, Gulvik CA, González JM (October 2014). "Master recyclers: features and functions of bacteria associated with phytoplankton blooms". Nature Reviews. Microbiology. 12 (10): 686–98. doi:10.1038/nrmicro3326. PMID25134618. S2CID26684717.
↑ Teeling H, Fuchs BM, Becher D, Klockow C, Gardebrecht A, Bennke CM, Kassabgy M, Huang S, Mann AJ, Waldmann J, Weber M, Klindworth A, Otto A, Lange J, Bernhardt J, Reinsch C, Hecker M, Peplies J, Bockelmann FD, Callies U, Gerdts G, Wichels A, Wiltshire KH, Glöckner FO, Schweder T, Amann R (May 2012). "Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom". Science. 336 (6081): 608–11. Bibcode:2012Sci...336..608T. doi:10.1126/science.1218344. PMID22556258. S2CID29249533.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.