
An osteoclast is a type of bone cell that breaks down bone tissue. This function is critical in the maintenance, repair, and remodeling of bones of the vertebral skeleton. The osteoclast disassembles and digests the composite of hydrated protein and mineral at a molecular level by secreting acid and a collagenase, a process known as bone resorption. This process also helps regulate the level of blood calcium.

Bisphosphonates are a class of drugs that prevent the loss of bone density, used to treat osteoporosis and similar diseases. They are the most commonly prescribed drugs used to treat osteoporosis. They are called bisphosphonates because they have two phosphonate groups. They are thus also called diphosphonates.

Alendronic acid, sold under the brand name Fosamax among others, is a bisphosphonate medication used to treat osteoporosis and Paget's disease of bone. It is taken by mouth. Use is often recommended together with vitamin D, calcium supplementation, and lifestyle changes.

Prenylation is the addition of hydrophobic molecules to a protein or a biomolecule. It is usually assumed that prenyl groups (3-methylbut-2-en-1-yl) facilitate attachment to cell membranes, similar to lipid anchors like the GPI anchor, though direct evidence of this has not been observed. Prenyl groups have been shown to be important for protein–protein binding through specialized prenyl-binding domains.
Farnesyl pyrophosphate (FPP), also known as farnesyl diphosphate (FDP), is an intermediate in the biosynthesis of terpenes and terpenoids such as sterols and carotenoids. It is also used in the synthesis of CoQ, as well as dehydrodolichol diphosphate.

The enzyme citrate synthase E.C. 2.3.3.1 ] exists in nearly all living cells and stands as a pace-making enzyme in the first step of the citric acid cycle. Citrate synthase is localized within eukaryotic cells in the mitochondrial matrix, but is encoded by nuclear DNA rather than mitochondrial. It is synthesized using cytoplasmic ribosomes, then transported into the mitochondrial matrix.
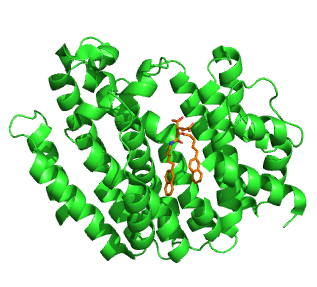
Squalene synthase (SQS) or farnesyl-diphosphate:farnesyl-diphosphate farnesyl transferase is an enzyme localized to the membrane of the endoplasmic reticulum. SQS participates in the isoprenoid biosynthetic pathway, catalyzing a two-step reaction in which two identical molecules of farnesyl pyrophosphate (FPP) are converted into squalene, with the consumption of NADPH. Catalysis by SQS is the first committed step in sterol synthesis, since the squalene produced is converted exclusively into various sterols, such as cholesterol, via a complex, multi-step pathway. SQS belongs to squalene/phytoene synthase family of proteins.
Purine metabolism refers to the metabolic pathways to synthesize and break down purines that are present in many organisms.

Camptothecin (CPT) is a topoisomerase inhibitor. It was discovered in 1966 by M. E. Wall and M. C. Wani in systematic screening of natural products for anticancer drugs. It was isolated from the bark and stem of Camptotheca acuminata, a tree native to China used in traditional Chinese medicine. It has been used clinically more recently in China for the treatment of gastrointestinal tumors. CPT showed anticancer activity in preliminary clinical trials, especially against breast, ovarian, colon, lung, and stomach cancers. However, it has low solubility and adverse effects have been reported when used therapeutically, so synthetic and medicinal chemists have developed numerous syntheses of camptothecin and various derivatives to increase the benefits of the chemical, with good results. Four CPT analogues have been approved and are used in cancer chemotherapy today: topotecan, irinotecan, belotecan, and trastuzumab deruxtecan. Camptothecin has also been found in other plants including Chonemorpha fragrans.
The discovery of an orally inactive peptide from snake venom established the important role of angiotensin converting enzyme (ACE) inhibitors in regulating blood pressure. This led to the development of captopril, the first ACE inhibitor. When the adverse effects of captopril became apparent new derivates were designed. Then after the discovery of two active sites of ACE: N-domain and C-domain, the development of domain-specific ACE inhibitors began.
In enzymology, a geranyltranstransferase is an enzyme that catalyzes the chemical reaction

Zaragozic acids are a family of natural products produced by fungi. The first characterized zaragozic acids, A, B, and C were isolated from an unidentified sterile fungal culture, Sporormiella intermedia, and L. elatius, respectively. just outside the European city Zaragoza, Spain on the Jalón river. This family of natural products possesses a unique 4,8-dioxabicyclo[3.2.1]octane core, and vary in their 1-alkyl and their 6-acyl side chains.
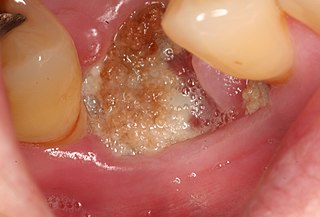
Medication-related osteonecrosis of the jaw is progressive death of the jawbone in a person exposed to a medication known to increase the risk of disease, in the absence of a previous radiation treatment. It may lead to surgical complication in the form of impaired wound healing following oral and maxillofacial surgery, periodontal surgery, or endodontic therapy.
Discovery and development of nucleoside and nucleotide reverse-transcriptase inhibitors began in the 1980s when the AIDS epidemic hit Western societies. NRTIs inhibit the reverse transcriptase (RT), an enzyme that controls the replication of the genetic material of the human immunodeficiency virus (HIV). The first NRTI was zidovudine, approved by the U.S. Food and Drug Administration (FDA) in 1987, which was the first step towards treatment of HIV. Six NRTI agents and one NtRTI have followed. The NRTIs and the NtRTI are analogues of endogenous 2´-deoxy-nucleoside and nucleotide. Drug-resistant viruses are an inevitable consequence of prolonged exposure of HIV-1 to anti-HIV drugs.
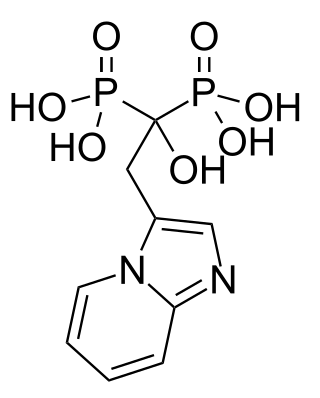
Minodronic acid is a third-generation bisphosphonate drug. It is approved for use in Japan for the treatment of osteoporosis. Its mechanism of action involves inhibition of farnesyl pyrophosphate synthase activity.
A steroidogenesis inhibitor, also known as a steroid biosynthesis inhibitor, is a type of drug which inhibits one or more of the enzymes that are involved in the process of steroidogenesis, the biosynthesis of endogenous steroids and steroid hormones. They may inhibit the production of cholesterol and other sterols, sex steroids such as androgens, estrogens, and progestogens, corticosteroids such as glucocorticoids and mineralocorticoids, and neurosteroids. They are used in the treatment of a variety of medical conditions that depend on endogenous steroids.

Lipase inhibitors belong to a drug class that is used as an antiobesity agent. Their mode of action is to inhibit gastric and pancreatic lipases, enzymes that play an important role in the digestion of dietary fat. Lipase inhibitors are classified in the ATC-classification system as A08AB . Numerous compounds have been either isolated from nature, semi-synthesized, or fully synthesized and then screened for their lipase inhibitory activity but the only lipase inhibitor on the market is orlistat . Lipase inhibitors have also shown anticancer activity, by inhibiting fatty acid synthase.

First described in 1965, the moenomycins are a family of phosphoglycolipid antibiotics, metabolites of the bacterial genus Streptomyces. Moenomycin A is the founding member of the antibiotic family with the majority discovered by the end of the late 1970s.
This article is about the discovery and development of 5α-reductase inhibitors (5-ARIs), also known as dihydrotestosterone (DHT) blockers.
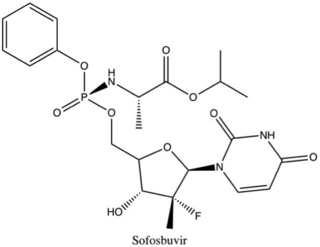
Non-structural protein 5B (NS5B) inhibitors are a class of direct-acting antivirals widely used in the treatment of chronic hepatitis C. Depending on site of action and chemical composition, NS5B inhibitors may be categorized into three classes—nucleoside active site inhibitors (NIs), non-nucleoside allosteric inhibitors, and pyrophosphate analogues. Subsequently, all three classes are then subclassified. All inhibit RNA synthesis by NS5B but at different stages/sites resulting in inability of viral RNA replication. Expression of direct-acting NS5B inhibitors does not take place in cells that are not infected by hepatitis C virus, which seems to be beneficial for this class of drugs.


