
In organic chemistry, an alkene, or olefin, is a hydrocarbon containing a carbon–carbon double bond. The double bond may be internal or in the terminal position. Terminal alkenes are also known as α-olefins.

In organic chemistry, ethers are a class of compounds that contain an ether group—an oxygen atom bonded to two organyl groups. They have the general formula R−O−R′, where R and R′ represent the organyl groups. Ethers can again be classified into two varieties: if the organyl groups are the same on both sides of the oxygen atom, then it is a simple or symmetrical ether, whereas if they are different, the ethers are called mixed or unsymmetrical ethers. A typical example of the first group is the solvent and anaesthetic diethyl ether, commonly referred to simply as "ether". Ethers are common in organic chemistry and even more prevalent in biochemistry, as they are common linkages in carbohydrates and lignin.
In chemistry, a nucleophilic substitution (SN) is a class of chemical reactions in which an electron-rich chemical species replaces a functional group within another electron-deficient molecule. The molecule that contains the electrophile and the leaving functional group is called the substrate.
Hydroboration–oxidation reaction is a two-step hydration reaction that converts an alkene into an alcohol. The process results in the syn addition of a hydrogen and a hydroxyl group where the double bond had been. Hydroboration–oxidation is an anti-Markovnikov reaction, with the hydroxyl group attaching to the less-substituted carbon. The reaction thus provides a more stereospecific and complementary regiochemical alternative to other hydration reactions such as acid-catalyzed addition and the oxymercuration–reduction process. The reaction was first reported by Herbert C. Brown in the late 1950s and it was recognized in his receiving the Nobel Prize in Chemistry in 1979.
In organic chemistry, an acyl chloride is an organic compound with the functional group −C(=O)Cl. Their formula is usually written R−COCl, where R is a side chain. They are reactive derivatives of carboxylic acids. A specific example of an acyl chloride is acetyl chloride, CH3COCl. Acyl chlorides are the most important subset of acyl halides.

A trimethylsilyl group (abbreviated TMS) is a functional group in organic chemistry. This group consists of three methyl groups bonded to a silicon atom [−Si(CH3)3], which is in turn bonded to the rest of a molecule. This structural group is characterized by chemical inertness and a large molecular volume, which makes it useful in a number of applications.
The Hiyama coupling is a palladium-catalyzed cross-coupling reaction of organosilanes with organic halides used in organic chemistry to form carbon–carbon bonds. This reaction was discovered in 1988 by Tamejiro Hiyama and Yasuo Hatanaka as a method to form carbon-carbon bonds synthetically with chemo- and regioselectivity. The Hiyama coupling has been applied to the synthesis of various natural products.
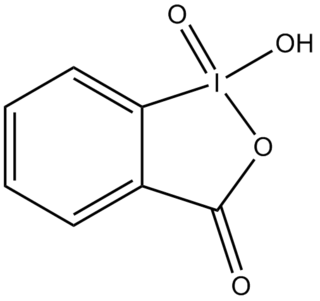
2-Iodoxybenzoic acid (IBX) is an organic compound used in organic synthesis as an oxidizing agent. This periodinane is especially suited to oxidize alcohols to aldehydes. IBX is most often prepared from 2-iodobenzoic acid and a strong oxidant such as potassium bromate and sulfuric acid, or more commonly, oxone. One of the main drawbacks of IBX is its limited solubility; IBX is insoluble in many common organic solvents. IBX is an impact- and heat-sensitive explosive (>200°C). Commercial IBX is stabilized by carboxylic acids such as benzoic acid and isophthalic acid.
Silyl ethers are a group of chemical compounds which contain a silicon atom covalently bonded to an alkoxy group. The general structure is R1R2R3Si−O−R4 where R4 is an alkyl group or an aryl group. Silyl ethers are usually used as protecting groups for alcohols in organic synthesis. Since R1R2R3 can be combinations of differing groups which can be varied in order to provide a number of silyl ethers, this group of chemical compounds provides a wide spectrum of selectivity for protecting group chemistry. Common silyl ethers are: trimethylsilyl (TMS), tert-butyldiphenylsilyl (TBDPS), tert-butyldimethylsilyl (TBS/TBDMS) and triisopropylsilyl (TIPS). They are particularly useful because they can be installed and removed very selectively under mild conditions.
Cyanogen bromide is the inorganic compound with the formula (CN)Br or BrCN. It is a colorless solid that is widely used to modify biopolymers, fragment proteins and peptides, and synthesize other compounds. The compound is classified as a pseudohalogen.
In organic chemistry the Brook rearrangement refers to any [1,n] carbon to oxygen silyl migration. The rearrangement was first observed in the late 1950s by Canadian chemist Adrian Gibbs Brook (1924–2013), after which the reaction is named. These migrations can be promoted in a number of different ways, including thermally, photolytically or under basic/acidic conditions. In the forward direction, these silyl migrations produce silyl ethers as products which is driven by the stability of the oxygen-silicon bond.

The Dakin oxidation (or Dakin reaction) is an organic redox reaction in which an ortho- or para-hydroxylated phenyl aldehyde (2-hydroxybenzaldehyde or 4-hydroxybenzaldehyde) or ketone reacts with hydrogen peroxide (H2O2) in base to form a benzenediol and a carboxylate. Overall, the carbonyl group is oxidised, whereas the H2O2 is reduced.

Organosilicon chemistry is the study of organometallic compounds containing carbon–silicon bonds, to which they are called organosilicon compounds. Most organosilicon compounds are similar to the ordinary organic compounds, being colourless, flammable, hydrophobic, and stable to air. Silicon carbide is an inorganic compound.
In organosilicon chemistry, silyl enol ethers are a class of organic compounds that share the common functional group R3Si−O−CR=CR2, composed of an enolate bonded to a silane through its oxygen end and an ethene group as its carbon end. They are important intermediates in organic synthesis.
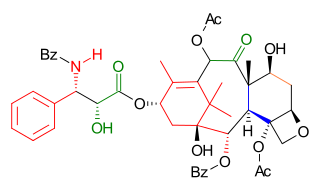
Wender Taxol total synthesis in organic chemistry describes a Taxol total synthesis by the group of Paul Wender at Stanford University published in 1997. This synthesis has much in common with the Holton Taxol total synthesis in that it is a linear synthesis starting from a naturally occurring compound with ring construction in the order A,B,C,D. The Wender effort is shorter by approximately 10 steps.

Phenylboronic acid or benzeneboronic acid, abbreviated as PhB(OH)2 where Ph is the phenyl group C6H5- and B(OH)2 is a boronic acid containing a phenyl substituent and two hydroxyl groups attached to boron. Phenylboronic acid is a white powder and is commonly used in organic synthesis. Boronic acids are mild Lewis acids which are generally stable and easy to handle, making them important to organic synthesis.

The Mukaiyama taxol total synthesis published by the group of Teruaki Mukaiyama of the Tokyo University of Science between 1997 and 1999 was the 6th successful taxol total synthesis. The total synthesis of Taxol is considered a hallmark in organic synthesis.
Electrophilic substitution of unsaturated silanes involves attack of an electrophile on an allyl- or vinylsilane. An allyl or vinyl group is incorporated at the electrophilic center after loss of the silyl group.
Electrophilic aromatic substitution (SEAr) is an organic reaction in which an atom that is attached to an aromatic system is replaced by an electrophile. Some of the most important electrophilic aromatic substitutions are aromatic nitration, aromatic halogenation, aromatic sulfonation, alkylation Friedel–Crafts reaction and acylation Friedel–Crafts reaction.
The Davis–Beirut reaction is N,N-bond forming heterocyclization that creates numerous types of 2H-indazoles and indazolones in both acidic and basic conditions The Davis–Beirut reaction is named after Mark Kurth and Makhluf Haddadin's respective universities; University of California, Davis and American University of Beirut, and is appealing because it uses inexpensive starting materials and does not require toxic metals.







