Related Research Articles
Nephrology is a specialty for both adult internal medicine and pediatric medicine that concerns the study of the kidneys, specifically normal kidney function and kidney disease, the preservation of kidney health, and the treatment of kidney disease, from diet and medication to renal replacement therapy. The word "renal" is an adjective meaning "relating to the kidneys", and its roots are French or late Latin. Whereas according to some opinions, "renal" and "nephro" should be replaced with "kidney" in scientific writings such as "kidney medicine" or "kidney replacement therapy", other experts have advocated preserving the use of renal and nephro as appropriate including in "nephrology" and "renal replacement therapy", respectively.

Autosomal dominant polycystic kidney disease (ADPKD) is one of the most common, life-threatening inherited human disorders and the most common hereditary kidney disease. It is associated with large interfamilial and intrafamilial variability, which can be explained to a large extent by its genetic heterogeneity and modifier genes. It is also the most common of the inherited cystic kidney diseases — a group of disorders with related but distinct pathogenesis, characterized by the development of renal cysts and various extrarenal manifestations, which in case of ADPKD include cysts in other organs, such as the liver, seminal vesicles, pancreas, and arachnoid membrane, as well as other abnormalities, such as intracranial aneurysms and dolichoectasias, aortic root dilatation and aneurysms, mitral valve prolapse, and abdominal wall hernias. Over 50% of patients with ADPKD eventually develop end stage kidney disease and require dialysis or kidney transplantation. ADPKD is estimated to affect at least one in every 1000 individuals worldwide, making this disease the most common inherited kidney disorder with a diagnosed prevalence of 1:2000 and incidence of 1:3000-1:8000 in a global scale.

Kidney dialysis is the process of removing excess water, solutes, and toxins from the blood in people whose kidneys can no longer perform these functions naturally. This is referred to as renal replacement therapy. The first successful dialysis was performed in 1943.

The nephron is the minute or microscopic structural and functional unit of the kidney. It is composed of a renal corpuscle and a renal tubule. The renal corpuscle consists of a tuft of capillaries called a glomerulus and a cup-shaped structure called Bowman's capsule. The renal tubule extends from the capsule. The capsule and tubule are connected and are composed of epithelial cells with a lumen. A healthy adult has 1 to 1.5 million nephrons in each kidney. Blood is filtered as it passes through three layers: the endothelial cells of the capillary wall, its basement membrane, and between the foot processes of the podocytes of the lining of the capsule. The tubule has adjacent peritubular capillaries that run between the descending and ascending portions of the tubule. As the fluid from the capsule flows down into the tubule, it is processed by the epithelial cells lining the tubule: water is reabsorbed and substances are exchanged ; first with the interstitial fluid outside the tubules, and then into the plasma in the adjacent peritubular capillaries through the endothelial cells lining that capillary. This process regulates the volume of body fluid as well as levels of many body substances. At the end of the tubule, the remaining fluid—urine—exits: it is composed of water, metabolic waste, and toxins.

Kidney failure, also known as end-stage renal disease (ESRD), is a medical condition in which the kidneys can no longer adequately filter waste products from the blood, functioning at less than 15% of normal levels. Kidney failure is classified as either acute kidney failure, which develops rapidly and may resolve; and chronic kidney failure, which develops slowly and can often be irreversible. Symptoms may include leg swelling, feeling tired, vomiting, loss of appetite, and confusion. Complications of acute and chronic failure include uremia, hyperkalemia, and volume overload. Complications of chronic failure also include heart disease, high blood pressure, and anaemia.

The glomerulus is a network of small blood vessels (capillaries) known as a tuft, located at the beginning of a nephron in the kidney. Each of the two kidneys contains about one million nephrons. The tuft is structurally supported by the mesangium, composed of intraglomerular mesangial cells. The blood is filtered across the capillary walls of this tuft through the glomerular filtration barrier, which yields its filtrate of water and soluble substances to a cup-like sac known as Bowman's capsule. The filtrate then enters the renal tubule of the nephron.

Alport syndrome is a genetic disorder affecting around 1 in 5,000-10,000 children, characterized by glomerulonephritis, end-stage kidney disease, and hearing loss. Alport syndrome can also affect the eyes, though the changes do not usually affect vision, except when changes to the lens occur in later life. Blood in urine is universal. Proteinuria is a feature as kidney disease progresses.

Diabetic nephropathy, also known as diabetic kidney disease, is the chronic loss of kidney function occurring in those with diabetes mellitus. Diabetic nephropathy is the leading causes of chronic kidney disease (CKD) and end-stage renal disease (ESRD) globally. The triad of protein leaking into the urine, rising blood pressure with hypertension and then falling renal function is common to many forms of CKD. Protein loss in the urine due to damage of the glomeruli may become massive, and cause a low serum albumin with resulting generalized body swelling (edema) so called nephrotic syndrome. Likewise, the estimated glomerular filtration rate (eGFR) may progressively fall from a normal of over 90 ml/min/1.73m2 to less than 15, at which point the patient is said to have end-stage renal disease. It usually is slowly progressive over years.

Hydronephrosis describes hydrostatic dilation of the renal pelvis and calyces as a result of obstruction to urine flow downstream. Alternatively, hydroureter describes the dilation of the ureter, and hydronephroureter describes the dilation of the entire upper urinary tract.
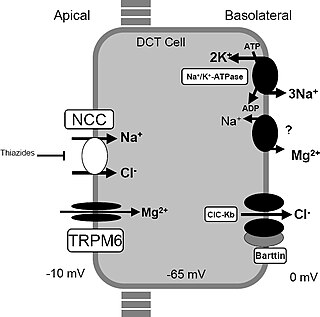
Gitelman syndrome (GS) is an autosomal recessive kidney tubule disorder characterized by low blood levels of potassium and magnesium, decreased excretion of calcium in the urine, and elevated blood pH. It is the most frequent hereditary salt-losing tubulopathy. Gitelman syndrome is caused by disease-causing variants on both alleles of the SLC12A3 gene. The SLC12A3 gene encodes the thiazide-sensitive sodium-chloride cotransporter, which can be found in the distal convoluted tubule of the kidney.
Thin basement membrane disease is, along with IgA nephropathy, the most common cause of hematuria without other symptoms. The only abnormal finding in this disease is a thinning of the basement membrane of the glomeruli in the kidneys. Its importance lies in the fact that it has a benign prognosis, with patients maintaining a normal kidney function throughout their lives.
Secondary hypertension is a type of hypertension which by definition is caused by an identifiable underlying primary cause. It is much less common than the other type, called essential hypertension, affecting only 5-10% of hypertensive patients. It has many different causes including endocrine diseases, kidney diseases, and tumors. The cause of secondary hypertension varies significantly with age. It also can be a side effect of many medications.

Renal tubular acidosis (RTA) is a medical condition that involves an accumulation of acid in the body due to a failure of the kidneys to appropriately acidify the urine. In renal physiology, when blood is filtered by the kidney, the filtrate passes through the tubules of the nephron, allowing for exchange of salts, acid equivalents, and other solutes before it drains into the bladder as urine. The metabolic acidosis that results from RTA may be caused either by insufficient secretion of hydrogen ions into the latter portions of the nephron or by failure to reabsorb sufficient bicarbonate ions from the filtrate in the early portion of the nephron. Although a metabolic acidosis also occurs in those with chronic kidney disease, the term RTA is reserved for individuals with poor urinary acidification in otherwise well-functioning kidneys. Several different types of RTA exist, which all have different syndromes and different causes. RTA is usually an incidental finding based on routine blood draws that show abnormal results. Clinically, patients may present with vague symptoms such as dehydration, mental status changes, or delayed growth in adolescents.

Cystic kidney disease refers to a wide range of hereditary, developmental, and acquired conditions and with the inclusion of neoplasms with cystic changes, over 40 classifications and subtypes have been identified. Depending on the disease classification, the presentation may be at birth, or much later into adult life. Cystic disease may involve one or both kidneys and may, or may not, occur in the presence of other anomalies. A higher incidence is found in males and prevalence increases with age. Renal cysts have been reported in more than 50% of patients over the age of 50. Typically, cysts grow up to 2.88 mm annually and may cause related pain and/or hemorrhage.
Familial renal disease is an uncommon cause of kidney failure in dogs and cats. Most causes are breed-related (familial) and some are inherited. Some are congenital. Renal dysplasia is a type of familial kidney disease characterized by abnormal cellular differentiation of kidney tissue. Dogs and cats with kidney disease caused by these diseases have the typical symptoms of kidney failure, including weight loss, loss of appetite, depression, and increased water consumption and urination. A list of familial kidney diseases by dog and cat breeds is found below.
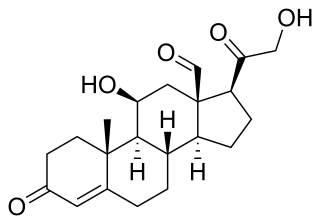
Pseudohypoaldosteronism (PHA) is a condition that mimics hypoaldosteronism.

Nephronophthisis is a genetic disorder of the kidneys which affects children. It is classified as a medullary cystic kidney disease. The disorder is inherited in an autosomal recessive fashion and, although rare, is the most common genetic cause of childhood kidney failure. It is a form of ciliopathy. Its incidence has been estimated to be 0.9 cases per million people in the United States, and 1 in 50,000 births in Canada.

Medullary sponge kidney is a congenital disorder of the kidneys characterized by cystic dilatation of the collecting tubules in one or both kidneys. Individuals with medullary sponge kidney are at increased risk for kidney stones and urinary tract infection (UTI). Patients with MSK typically pass twice as many stones per year as do other stone formers without MSK. While having a low morbidity rate, as many as 10% of patients with MSK have an increased risk of morbidity associated with frequent stones and UTIs. While many patients report increased chronic kidney pain, the source of the pain, when a UTI or blockage is not present, is unclear at this time. Renal colic is present in 55% of patients. Women with MSK experience more stones, UTIs, and complications than men. MSK was previously believed not to be hereditary but there is more evidence coming forth that may indicate otherwise.
Polycystic kidney disease is a genetic disorder in which the renal tubules become structurally abnormal, resulting in the development and growth of multiple cysts within the kidney. These cysts may begin to develop in utero, in infancy, in childhood, or in adulthood. Cysts are non-functioning tubules filled with fluid pumped into them, which range in size from microscopic to enormous, crushing adjacent normal tubules and eventually rendering them non-functional as well.

Thomas Martin Barratt was a British paediatrician and professor of paediatric nephrology. Barratt was most notable for developing a specialist service for children with kidney diseases in Britain, bringing peritoneal dialysis, haemodialysis, and later renal transplantation to ever younger children. Barratt was an early advocate for multidisciplinary care and developed a model that was later taken up by many other specialist centres across the world. His research led to a new treatments for many types of childhood kidney diseases., and for research into childhood Nephrotic syndrome and Hemolytic-uremic syndrome.
References
- ↑ Bissler, John J.; Siroky, Brian J.; Yin, Hong (October 2010). "Glomerulocystic kidney disease". Pediatric Nephrology. 25 (10): 2049–2059. doi:10.1007/s00467-009-1416-2. PMC 2923715 . PMID 20091054.
- ↑ Miyazaki, K; Miyazaki, M; Yoshizuka, N; Sasaki, O (May 2002). "Glomerulocystic kidney disease (GCKD) associated with Henoch-Schoenlein purpura: a case report and a review of adult cases of GCKD". Clinical Nephrology. 57 (5). Dustri-Verlag: 386–391.
- 1 2 3 4 Sahay, M., & Gowrishankar, S. (2010). Glomerulocystic disease. NDT plus, 3(4), 349–350. doi:10.1093/ndtplus/sfq048
- ↑ Bissler, John J.; Siroky, Brian J.; Yin, Hong (October 2010). "Glomerulocystic kidney disease". Pediatric Nephrology. 25 (10): 2049–2059. doi:10.1007/s00467-009-1416-2. PMC 2923715 . PMID 20091054.
- ↑ Sung MH. (2013) Transcriptional Reprogramming. In: Dubitzky W., Wolkenhauer O., Cho KH., Yokota H. (eds) Encyclopedia of Systems Biology. Springer, New York, NY
- 1 2 3 4 5 6 7 Rosanna Gusmano, Gianluca Caridi, Monica Marini, Francesco Perfumo, Gian Marco Ghiggeri, Giorgio Piaggio, Isabella Ceccherini, Marco Seri, Glomerulocystic kidney disease in a family, Nephrology Dialysis Transplantation, Volume 17, Issue 5, May 2002, Pages 813–818, https://doi.org/10.1093/ndt/17.5.813
- 1 2 Bissler, J. J., Siroky, B. J., & Yin, H. (2010). Glomerulocystic kidney disease. Pediatric nephrology (Berlin, Germany), 25(10), 2049–2059. doi:10.1007/s00467-009-1416-2
- ↑ Bissler, John J.; Siroky, Brian J.; Yin, Hong (October 2010). "Glomerulocystic kidney disease". Pediatric Nephrology. 25 (10): 2049–2059. doi:10.1007/s00467-009-1416-2. PMC 2923715 . PMID 20091054.
- 1 2 Bissler, J.J., Siroky, B.J. & Yin, H. Pediatr Nephrol (2010) 25: 2049. https://doi.org/10.1007/s00467-009-1416-2
- 1 2 Srivastava, R. N., & Bagga, A. (2016). Pediatric nephrology. New Delhi: Jaypee Brothers Medical Publishers (P) LTD.
- ↑ Oliva, M., Hsing, J., Rybicki, F. et al. Abdom Imaging (2003) 28: 889. https://doi.org/10.1007/s00261-003-0024-z
- ↑ Lundquist, A. L. (2019, January 23). "Hereditary Renal Cystic Diseases: Glomerulocystic Kidney Disease"