Autosomal dominant polycystic kidney disease (ADPKD) is one of the most common, life-threatening inherited human disorders and the most common hereditary kidney disease. It is associated with large interfamilial and intrafamilial variability, which can be explained to a large extent by its genetic heterogeneity and modifier genes. It is also the most common of the inherited cystic kidney diseases — a group of disorders with related but distinct pathogenesis, characterized by the development of renal cysts and various extrarenal manifestations, which in case of ADPKD include cysts in other organs, such as the liver, seminal vesicles, pancreas, and arachnoid membrane, as well as other abnormalities, such as intracranial aneurysms and dolichoectasias, aortic root dilatation and aneurysms, mitral valve prolapse, and abdominal wall hernias. Over 50% of patients with ADPKD eventually develop end stage kidney disease and require dialysis or kidney transplantation. ADPKD is estimated to affect at least one in every 1000 individuals worldwide, making this disease the most common inherited kidney disorder with a diagnosed prevalence of 1:2000 and incidence of 1:3000-1:8000 in a global scale.

Caroli disease is a rare inherited disorder characterized by cystic dilatation of the bile ducts within the liver. There are two patterns of Caroli disease: focal or simple Caroli disease consists of abnormally widened bile ducts affecting an isolated portion of liver. The second form is more diffuse, and when associated with portal hypertension and congenital hepatic fibrosis, is often referred to as "Caroli syndrome". The underlying differences between the two types are not well understood. Caroli disease is also associated with liver failure and polycystic kidney disease. The disease affects about one in 1,000,000 people, with more reported cases of Caroli syndrome than of Caroli disease.

Tuberous sclerosis complex (TSC) is a rare multisystem autosomal dominant genetic disease that causes non-cancerous tumours to grow in the brain and on other vital organs such as the kidneys, heart, liver, eyes, lungs and skin. A combination of symptoms may include seizures, intellectual disability, developmental delay, behavioral problems, skin abnormalities, lung disease, and kidney disease.

Kidney disease, or renal disease, technically referred to as nephropathy, is damage to or disease of a kidney. Nephritis is an inflammatory kidney disease and has several types according to the location of the inflammation. Inflammation can be diagnosed by blood tests. Nephrosis is non-inflammatory kidney disease. Nephritis and nephrosis can give rise to nephritic syndrome and nephrotic syndrome respectively. Kidney disease usually causes a loss of kidney function to some degree and can result in kidney failure, the complete loss of kidney function. Kidney failure is known as the end-stage of kidney disease, where dialysis or a kidney transplant is the only treatment option.

Alport syndrome is a genetic disorder affecting around 1 in 5,000-10,000 children, characterized by glomerulonephritis, end-stage kidney disease, and hearing loss. Alport syndrome can also affect the eyes, though the changes do not usually affect vision, except when changes to the lens occur in later life. Blood in urine is universal. Proteinuria is a feature as kidney disease progresses.

Cystinuria is an inherited autosomal recessive disease characterized by high concentrations of the amino acid cystine in the urine, leading to the formation of cystine stones in the kidneys, ureters, and bladder. It is a type of aminoaciduria. "Cystine", not "cysteine," is implicated in this disease; the former is a dimer of the latter.
Potter sequence is the atypical physical appearance of a baby due to oligohydramnios experienced when in the uterus. It includes clubbed feet, pulmonary hypoplasia and cranial anomalies related to the oligohydramnios. Oligohydramnios is the decrease in amniotic fluid volume sufficient to cause deformations in morphogenesis of the baby.

Barakat syndrome is a rare disease characterized by hypoparathyroidism, sensorineural deafness and renal disease, and hence also known as HDR syndrome. It was first described by Amin J. Barakat et al. in 1977.
Medullary cystic kidney disease (MCKD) is an autosomal dominant kidney disorder characterized by tubulointerstitial sclerosis leading to end-stage renal disease. Because the presence of cysts is neither an early nor a typical diagnostic feature of the disease, and because at least 4 different gene mutations may give rise to the condition, the name autosomal dominant tubulointerstitial kidney disease (ADTKD) has been proposed, to be appended with the underlying genetic variant for a particular individual. Importantly, if cysts are found in the medullary collecting ducts they can result in a shrunken kidney, unlike that of polycystic kidney disease. There are two known forms of medullary cystic kidney disease, mucin-1 kidney disease 1 (MKD1) and mucin-2 kidney disease/uromodulin kidney disease (MKD2). A third form of the disease occurs due to mutations in the gene encoding renin (ADTKD-REN), and has formerly been known as familial juvenile hyperuricemic nephropathy type 2.
Papillorenal syndrome is an autosomal dominant genetic disorder marked by underdevelopment (hypoplasia) of the kidney and colobomas of the optic nerve.
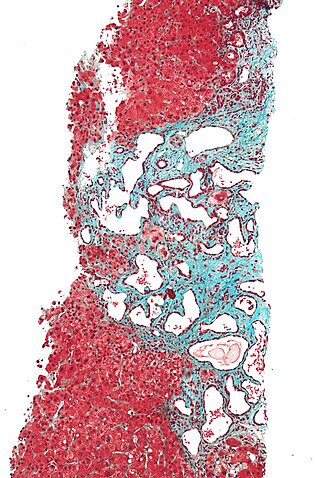
Polycystic liver disease (PLD) usually describes the presence of multiple cysts scattered throughout normal liver tissue. PLD is commonly seen in association with autosomal-dominant polycystic kidney disease, with a prevalence of 1 in 400 to 1000, and accounts for 8–10% of all cases of end-stage renal disease. The much rarer autosomal-dominant polycystic liver disease will progress without any kidney involvement.

Nephronophthisis is a genetic disorder of the kidneys which affects children. It is classified as a medullary cystic kidney disease. The disorder is inherited in an autosomal recessive fashion and, although rare, is the most common genetic cause of childhood kidney failure. It is a form of ciliopathy. Its incidence has been estimated to be 0.9 cases per million people in the United States, and 1 in 50,000 births in Canada.

Polycystin 1 is a protein that in humans is encoded by the PKD1 gene. Mutations of PKD1 are associated with most cases of autosomal dominant polycystic kidney disease, a severe hereditary disorder of the kidneys characterised by the development of renal cysts and severe kidney dysfunction.

Medullary sponge kidney is a congenital disorder of the kidneys characterized by cystic dilatation of the collecting tubules in one or both kidneys. Individuals with medullary sponge kidney are at increased risk for kidney stones and urinary tract infection (UTI). Patients with MSK typically pass twice as many stones per year as do other stone formers without MSK. While having a low morbidity rate, as many as 10% of patients with MSK have an increased risk of morbidity associated with frequent stones and UTIs. While many patients report increased chronic kidney pain, the source of the pain, when a UTI or blockage is not present, is unclear at this time. Renal colic is present in 55% of patients. Women with MSK experience more stones, UTIs, and complications than men. MSK was previously believed not to be hereditary but there is more evidence coming forth that may indicate otherwise.
Polycystic kidney disease is a genetic disorder in which the renal tubules become structurally abnormal, resulting in the development and growth of multiple cysts within the kidney. These cysts may begin to develop in utero, in infancy, in childhood, or in adulthood. Cysts are non-functioning tubules filled with fluid pumped into them, which range in size from microscopic to enormous, crushing adjacent normal tubules and eventually rendering them non-functional as well.
Autosomal recessive polycystic kidney disease (ARPKD) is the recessive form of polycystic kidney disease. It is associated with a group of congenital fibrocystic syndromes. Mutations in the PKHD1 cause ARPKD.

Lixivaptan (VPA-985) is an orally-active, non-peptide, selective vasopressin 2 receptor antagonist being developed as an investigational drug by Palladio Biosciences, Inc. (Palladio), a subsidiary of Centessa Pharmaceuticals plc. As of December 2021, lixivaptan is in Phase III clinical development for the treatment of Autosomal dominant polycystic kidney disease (ADPKD), the most common form of polycystic kidney disease. The U.S. Food and Drug Administration (FDA) has granted orphan drug designation to lixivaptan for the treatment of ADPKD.
Glomerulocystic kidney disease (GCKD) is a cystic disorder of the kidneys. GCKD involves cystic dilation of Bowman's capsule. It can occur with or without congenital abnormality.
Polycystic kidney disease 3 (autosomal dominant) is a protein that in humans is encoded by the PKD3 gene.
COACH syndrome, also known as Joubert syndrome with hepatic defect, is a rare autosomal recessive genetic disease. The name is an acronym of the defining signs: cerebellar vermis aplasia, oligophrenia, congenital ataxia, coloboma and hepatic fibrosis. The condition is associated with moderate intellectual disability. It falls under the category of a Joubart Syndrome-related disorder (JSRD).