
Estrone (E1), also spelled oestrone, is a steroid, a weak estrogen, and a minor female sex hormone. It is one of three major endogenous estrogens, the others being estradiol and estriol. Estrone, as well as the other estrogens, are synthesized from cholesterol and secreted mainly from the gonads, though they can also be formed from adrenal androgens in adipose tissue. Relative to estradiol, both estrone and estriol have far weaker activity as estrogens. Estrone can be converted into estradiol, and serves mainly as a precursor or metabolic intermediate of estradiol. It is both a precursor and metabolite of estradiol.

Estriol (E3), also spelled oestriol, is a steroid, a weak estrogen, and a minor female sex hormone. It is one of three major endogenous estrogens, the others being estradiol and estrone. Levels of estriol in women who are not pregnant are almost undetectable. However, during pregnancy, estriol is synthesized in very high quantities by the placenta and is the most produced estrogen in the body by far, although circulating levels of estriol are similar to those of other estrogens due to a relatively high rate of metabolism and excretion. Relative to estradiol, both estriol and estrone have far weaker activity as estrogens.
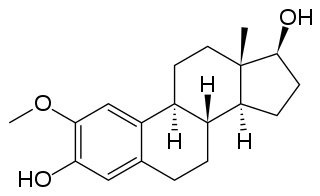
2-Methoxyestradiol is a natural metabolite of estradiol and 2-hydroxyestradiol (2-OHE2). It is specifically the 2-methyl ether of 2-hydroxyestradiol. 2-Methoxyestradiol prevents the formation of new blood vessels that tumors need in order to grow (angiogenesis), hence it is an angiogenesis inhibitor. It also acts as a vasodilator and induces apoptosis in some cancer cell lines. 2-Methoxyestradiol is derived from estradiol, although it interacts poorly with the estrogen receptors. However, it retains activity as a high-affinity agonist of the G protein-coupled estrogen receptor (GPER).

X-linked ichthyosis is a skin condition caused by the hereditary deficiency of the steroid sulfatase (STS) enzyme that affects 1 in 2000 to 1 in 6000 males. XLI manifests with dry, scaly skin and is due to deletions or mutations in the STS gene. XLI can also occur in the context of larger deletions causing contiguous gene syndromes. Treatment is largely aimed at alleviating the skin symptoms. The term is from the Ancient Greek 'ichthys' meaning 'fish'.

Tibolone, sold under the brand name Livial among others, is a medication which is used in menopausal hormone therapy and in the treatment of postmenopausal osteoporosis and endometriosis. The medication is available alone and is not formulated or used in combination with other medications. It is taken by mouth.

Steroid sulfatase (STS), or steryl-sulfatase, formerly known as arylsulfatase C, is a sulfatase enzyme involved in the metabolism of steroids. It is encoded by the STS gene.

Megestrol acetate (MGA), sold under the brand name Megace among others, is a progestin medication which is used mainly as an appetite stimulant to treat wasting syndromes such as cachexia. It is also used to treat breast cancer and endometrial cancer, and has been used in birth control. Megestrol acetate is generally formulated alone, although it has been combined with estrogens in birth control formulations. It is usually taken by mouth.

Dienogest, sold under the brand name Visanne among others, is a progestin medication which is used in birth control pills and in the treatment of endometriosis. It is also used in menopausal hormone therapy and to treat heavy periods. Dienogest is available both alone and in combination with estrogens. It is taken by mouth.

Estrone sulfate, also known as E1S, E1SO4 and estrone 3-sulfate, is a natural, endogenous steroid and an estrogen ester and conjugate.
Hormone replacement therapy (HRT), also known as menopausal hormone therapy or postmenopausal hormone therapy, is a form of hormone therapy used to treat symptoms associated with female menopause. Effects of menopause can include symptoms such as hot flashes, accelerated skin aging, vaginal dryness, decreased muscle mass, and complications such as osteoporosis, sexual dysfunction, and vaginal atrophy. They are mostly caused by low levels of female sex hormones that occur during menopause.

Michael J. Reed was a British chemist who held the position of professor of steroid biochemistry at Imperial College, London.
A steroidogenesis inhibitor, also known as a steroid biosynthesis inhibitor, is a type of drug which inhibits one or more of the enzymes that are involved in the process of steroidogenesis, the biosynthesis of endogenous steroids and steroid hormones. They may inhibit the production of cholesterol and other sterols, sex steroids such as androgens, estrogens, and progestogens, corticosteroids such as glucocorticoids and mineralocorticoids, and neurosteroids. They are used in the treatment of a variety of medical conditions that depend on endogenous steroids.

Estradiol sulfamate, or estradiol-3-O-sulfamate, is a steroid sulfatase (STS) inhibitor which is under development for the treatment of endometriosis. It is the C3 sulfamate ester of estradiol, and was originally thought to be a prodrug of estradiol.

Estrone sulfamate, or estrone-3-O-sulfamate, is a steroid sulfatase (STS) inhibitor which has not yet been marketed. It is the C3 sulfamate ester of the estrogen estrone. Unlike other estrogen esters however, EMATE is not an effective prodrug of estrogens. A closely related compound is estradiol sulfamate (E2MATE), which is extensively metabolized into EMATE and has similar properties to it.

EC508, also known as estradiol 17β-(1- -L-proline), is an estrogen which is under development by Evestra for use in menopausal hormone therapy and as a hormonal contraceptive for the prevention of pregnancy in women. It is an orally active estrogen ester – specifically, a C17β sulfonamide–proline ester of the natural and bioidentical estrogen estradiol – and acts as a prodrug of estradiol in the body. However, unlike oral estradiol and conventional oral estradiol esters such as estradiol valerate, EC508 undergoes little or no first-pass metabolism, has high oral bioavailability, and does not have disproportionate estrogenic effects in the liver. As such, it has a variety of desirable advantages over oral estradiol, similarly to parenteral estradiol, but with the convenience of oral administration. EC508 is a candidate with the potential to replace not only oral estradiol in clinical practice, but also ethinylestradiol in oral contraceptives. Evestra intends to seek Investigational New Drug status for EC508 in the second quarter of 2018.

Estriol sulfamate, or estriol 3-O-sulfamate, is a synthetic estrogen and estrogen ester which was never marketed. It is the C3 sulfamate ester of estriol. The drug shows substantially improved oral estrogenic potency relative to estriol in rats but without an increase in hepatic estrogenic potency. However, the closely related compound estradiol sulfamate (E2MATE) failed to show estrogenic activity in humans, which is due to the fact that it is additionally a highly potent inhibitor of steroid sulfatase which regulates the estrogenicity of such compounds and thus it prevents its own bioactivation into estradiol.

The pharmacology of estradiol, an estrogen medication and naturally occurring steroid hormone, concerns its pharmacodynamics, pharmacokinetics, and various routes of administration.

Barry Victor Lloyd Potter MAE FMedSci is a British chemist, who is Professor of Medicinal & Biological Chemistry at the University of Oxford, Wellcome Trust Senior Investigator and a Fellow of University College, Oxford.

2-Methoxyestradiol disulfamate is a synthetic, oral active anti-cancer medication which was previously under development for potential clinical use. It has improved potency, low metabolism, and good pharmacokinetic properties relative to 2-methoxyestradiol (2-MeO-E2). It is also a potent inhibitor of steroid sulfatase, the enzyme that catalyzes the desulfation of steroids such as estrone sulfate and dehydroepiandrosterone sulfate (DHEA-S).

Linustedastat is a 17β-hydroxysteroid dehydrogenase 1 inhibitor which is under development for the treatment of endometriosis. It is a steroidal compound derived from estrone and works by preventing the formation of the more potent estrogen estradiol from the minimally active precursor estrone. This in turn results in antiestrogenic effects that may be useful in the treatment of estrogen-dependent conditions. As of November 2023, the drug is in phase 2 clinical trials for endometriosis. It is also under preclinical investigation for treatment of breast cancer and endometrial cancer.
