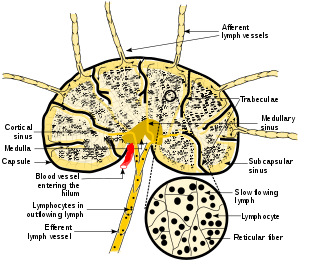
A lymph node, or lymph gland, is a kidney-shaped organ of the lymphatic system and the adaptive immune system. A large number of lymph nodes are linked throughout the body by the lymphatic vessels. They are major sites of lymphocytes that include B and T cells. Lymph nodes are important for the proper functioning of the immune system, acting as filters for foreign particles including cancer cells, but have no detoxification function.

Infectious mononucleosis, also known as glandular fever, is an infection usually caused by the Epstein–Barr virus (EBV). Most people are infected by the virus as children, when the disease produces few or no symptoms. In young adults, the disease often results in fever, sore throat, enlarged lymph nodes in the neck, and fatigue. Most people recover in two to four weeks; however, feeling tired may last for months. The liver or spleen may also become swollen, and in less than one percent of cases splenic rupture may occur.

Lymphogranuloma venereum is a sexually transmitted infection caused by the invasive serovars L1, L2, L2a, L2b, or L3 of Chlamydia trachomatis.
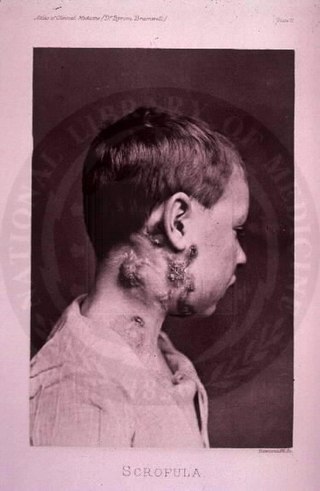
The disease mycobacterial cervical lymphadenitis, also known as scrofula and historically as king's evil, involves a lymphadenitis of the cervical lymph nodes associated with tuberculosis as well as nontuberculous (atypical) mycobacteria.

Castlemandisease (CD) describes a group of rare lymphoproliferative disorders that involve enlarged lymph nodes, and a broad range of inflammatory symptoms and laboratory abnormalities. Whether Castleman disease should be considered an autoimmune disease, cancer, or infectious disease is currently unknown.

Lymphadenopathy or adenopathy is a disease of the lymph nodes, in which they are abnormal in size or consistency. Lymphadenopathy of an inflammatory type is lymphadenitis, producing swollen or enlarged lymph nodes. In clinical practice, the distinction between lymphadenopathy and lymphadenitis is rarely made and the words are usually treated as synonymous. Inflammation of the lymphatic vessels is known as lymphangitis. Infectious lymphadenitis affecting lymph nodes in the neck is often called scrofula.
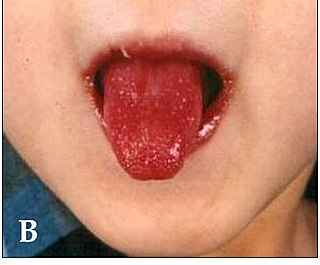
Kawasaki disease is a syndrome of unknown cause that results in a fever and mainly affects children under 5 years of age. It is a form of vasculitis, where medium-sized blood vessels become inflamed throughout the body. The fever typically lasts for more than five days and is not affected by usual medications. Other common symptoms include large lymph nodes in the neck, a rash in the genital area, lips, palms, or soles of the feet, and red eyes. Within three weeks of the onset, the skin from the hands and feet may peel, after which recovery typically occurs. The disease is the leading cause of acquired heart disease in children in developed countries, which include the formation of coronary artery aneurysms and myocarditis.
Talaromycosis is a fungal infection that presents with painless skin lesions of the face and neck, as well as an associated fever, anaemia, and enlargement of the lymph glands and liver.
Persistent generalized lymphadenopathy (PGL) is enlarged, painless, non-tender lymph nodes occurring in a couple of different areas for more than three to six months for which no other reason can be found. To expand, the common site where PGL occurs is within the head and neck region; parotid gland alterations and nasopharyngeal lymphatic tissue enlargement are often frequent comorbidities of Persistent generalized lymphadenopathy. Due to lymphoproliferation in the intraglandular lymphoid tissue, obstruction within the epithelium results in cystic expansion, which is the cause of cystic parotid lesions found in PGL. This condition frequently occurs in people in the latency period of HIV/AIDS.
Lymphoid hyperplasia is the rapid proliferation of normal lymphocytic cells that resemble lymph tissue which may occur with bacterial or viral infections. The growth is termed hyperplasia which may result in enlargement of various tissue including an organ, or cause a cutaneous lesion.

Rosai–Dorfman disease, also known as sinus histiocytosis with massive lymphadenopathy or sometimes as Destombes–Rosai–Dorfman disease, is a rare disorder of unknown cause that is characterized by abundant histiocytes in the lymph nodes or other locations throughout the body.
Diffuse infiltrative lymphocytosis syndrome (DILS) is a rare multi-system complication of HIV believed to occur secondary to an abnormal persistence of the initial CD8+ T cell expansion that regularly occurs in an HIV infection. This persistent CD8+ T cell expansion occurs in the setting of a low CD4+/CD8+ T cell ratio and ultimately invades and destroys tissues and organs resulting in the various complications of DILS. DILS classically presents with bilateral salivary gland enlargement (parotitis), cervical lymphadenopathy, and sicca symptoms such as xerophthalmia and xerostomia, but it may also involve the lungs, nervous system, kidneys, liver, digestive tract, and muscles. Once suspected, current diagnostic workups include (1) confirming HIV infection, (2) confirming six or greater months of characteristic signs and symptoms, (3) confirming organ infiltration by CD8+ T cells, and (4) exclusion of other autoimmune conditions. Once the diagnosis of DILS is confirmed, management includes highly active antiretroviral therapy (HAART) and as-needed steroids. With proper treatment, the overall prognosis of DILS is favorable.
Necrotizing vasculitis, also called systemic necrotizing vasculitus, is a category of vasculitis, comprising vasculitides that present with necrosis. Examples include giant cell arteritis, microscopic polyangiitis, and granulomatosis with polyangiitis. ICD-10 uses the variant "necrotizing vasculopathy". ICD-9, while classifying these conditions together, does not use a dedicated phrase, instead calling them "polyarteritis nodosa and allied conditions".

Cervical lymphadenopathy refers to lymphadenopathy of the cervical lymph nodes. The term lymphadenopathy strictly speaking refers to disease of the lymph nodes, though it is often used to describe the enlargement of the lymph nodes. Similarly, the term lymphadenitis refers to inflammation of a lymph node, but often it is used as a synonym of lymphadenopathy.
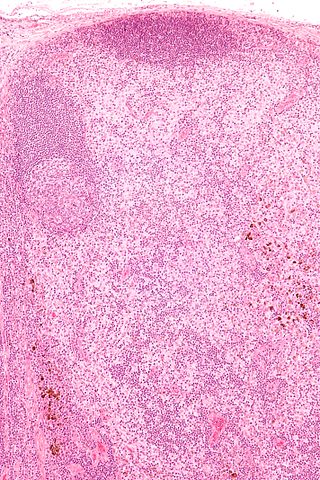
In pathology, dermatopathic lymphadenopathy, is lymph node pathology due to skin disease.

Cat-scratch disease (CSD) or felinosis is an infectious disease that most often results from a scratch or bite of a cat. Symptoms typically include a non-painful bump or blister at the site of injury and painful and swollen lymph nodes. People may feel tired, have a headache, or a fever. Symptoms typically begin within 3–14 days following infection.
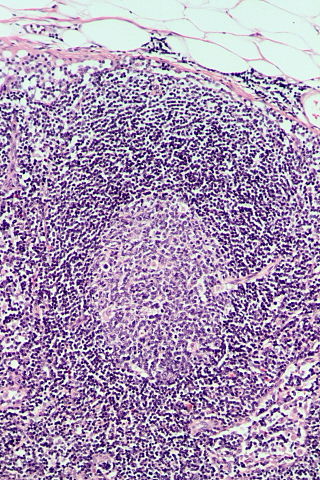
Follicular hyperplasia (FH) is a type of lymphoid hyperplasia and is classified as a lymphadenopathy, which means a disease of the lymph nodes. It is caused by a stimulation of the B cell compartment and by abnormal cell growth of secondary follicles. This typically occurs in the cortex without disrupting the lymph node capsule. The follicles are pathologically polymorphous, are often contrasting and varying in size and shape. Follicular hyperplasia is distinguished from follicular lymphoma in its polyclonality and lack of bcl-2 protein expression, whereas follicular lymphoma is monoclonal, and expresses bcl-2.
Unicentric Castleman disease is a subtype of Castleman disease, a group of lymphoproliferative disorders characterized by lymph node enlargement, characteristic features on microscopic analysis of enlarged lymph node tissue, and a range of symptoms and clinical findings.
Idiopathic multicentric Castleman disease (iMCD) is a subtype of Castleman disease (also known as giant lymph node hyperplasia, lymphoid hamartoma, or angiofollicular lymph node hyperplasia), a group of lymphoproliferative disorders characterized by lymph node enlargement, characteristic features on microscopic analysis of enlarged lymph node tissue, and a range of symptoms and clinical findings.
Epstein–Barr virus–associated lymphoproliferative diseases are a group of disorders in which one or more types of lymphoid cells, i.e. B cells, T cells, NK cells, and histiocytic-dendritic cells, are infected with the Epstein–Barr virus (EBV). This causes the infected cells to divide excessively, and is associated with the development of various non-cancerous, pre-cancerous, and cancerous lymphoproliferative disorders (LPDs). These LPDs include the well-known disorder occurring during the initial infection with the EBV, infectious mononucleosis, and the large number of subsequent disorders that may occur thereafter. The virus is usually involved in the development and/or progression of these LPDs although in some cases it may be an "innocent" bystander, i.e. present in, but not contributing to, the disease.
