
Say–Meyer syndrome is a rare X-linked genetic disorder that is mostly characterized as developmental delay. It is one of the rare causes of short stature. It is closely related with trigonocephaly. People with Say–Meyer syndrome have impaired growth, deficits in motor skills development and mental state.

The skull is a bone protective cavity for the brain. The skull is composed of three types of bone: cranial bones, facial bones, and ear ossicles. Two parts are more prominent: the cranium and the mandible. In humans, these two parts are the neurocranium (braincase) and the viscerocranium that includes the mandible as its largest bone. The skull forms the anterior-most portion of the skeleton and is a product of cephalisation—housing the brain, and several sensory structures such as the eyes, ears, nose, and mouth. In humans, these sensory structures are part of the facial skeleton.

A fontanelle is an anatomical feature of the infant human skull comprising soft membranous gaps (sutures) between the cranial bones that make up the calvaria of a fetus or an infant. Fontanelles allow for stretching and deformation of the neurocranium both during birth and later as the brain expands faster than the surrounding bone can grow. Premature complete ossification of the sutures is called craniosynostosis.

Plagiocephaly, also known as flat head syndrome, is a condition characterized by an asymmetrical distortion of the skull. A mild and widespread form is characterized by a flat spot on the back or one side of the head caused by remaining in a supine position for prolonged periods.
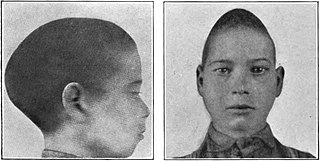
Scaphocephaly, or sagittal craniosynostosis, is a type of cephalic disorder which occurs when there is a premature fusion of the sagittal suture. Premature closure results in limited lateral expansion of the skull resulting in a characteristic long, narrow head. The skull base is typically spared.
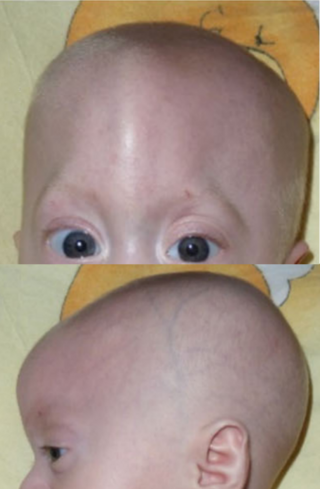
Trigonocephaly is a congenital condition due to premature fusion of the metopic suture, leading to a triangular forehead. The premature merging of the two frontal bones leads to transverse growth restriction and parallel growth expansion. It may occur as one component of a syndrome together with other abnormalities, or in isolated form. The term is from the Greek trigonon, "triangle", and kephale, "head".

Crouzon syndrome is an autosomal dominant genetic disorder known as a branchial arch syndrome. Specifically, this syndrome affects the first branchial arch, which is the precursor of the maxilla and mandible. Because the branchial arches are important developmental features in a growing embryo, disturbances in their development create lasting and widespread effects. The syndrome is caused by a mutation in a gene on chromosome 10 that controls the body's production of fibroblast growth factor receptor 2 (FGFR2).

Apert syndrome is a form of acrocephalosyndactyly, a congenital disorder characterized by malformations of the skull, face, hands and feet. It is classified as a branchial arch syndrome, affecting the first branchial arch, the precursor of the maxilla and mandible. Disturbances in the development of the branchial arches in fetal development create lasting and widespread effects.

Craniosynostosis is a condition in which one or more of the fibrous sutures in a young infant's skull prematurely fuses by turning into bone (ossification), thereby changing the growth pattern of the skull. Because the skull cannot expand perpendicular to the fused suture, it compensates by growing more in the direction parallel to the closed sutures. Sometimes the resulting growth pattern provides the necessary space for the growing brain, but results in an abnormal head shape and abnormal facial features. In cases in which the compensation does not effectively provide enough space for the growing brain, craniosynostosis results in increased intracranial pressure leading possibly to visual impairment, sleeping impairment, eating difficulties, or an impairment of mental development combined with a significant reduction in IQ.

Saethre–Chotzen syndrome (SCS), also known as acrocephalosyndactyly type III, is a rare congenital disorder associated with craniosynostosis. This affects the shape of the head and face, resulting in a cone-shaped head and an asymmetrical face. Individuals with SCS also have droopy eyelids (ptosis), widely spaced eyes (hypertelorism), and minor abnormalities of the hands and feet (syndactyly). Individuals with more severe cases of SCS may have mild to moderate intellectual or learning disabilities. Depending on the level of severity, some individuals with SCS may require some form of medical or surgical intervention. Most individuals with SCS live fairly normal lives, regardless of whether medical treatment is needed or not.
Craniofacial surgery is a surgical subspecialty that deals with congenital and acquired deformities of the head, skull, face, neck, jaws and associated structures. Although craniofacial treatment often involves manipulation of bone, craniofacial surgery is not tissue-specific; craniofacial surgeons deal with bone, skin, nerve, muscle, teeth, and other related anatomy.

Carpenter syndrome, also called acrocephalopolysyndactyly type II, is an extremely rare autosomal recessive congenital disorder characterized by craniofacial malformations, obesity, syndactyly, and polydactyly. Acrocephalopolysyndactyly is a variation of acrocephalosyndactyly that presents with polydactyly.

The sagittal suture, also known as the interparietal suture and the sutura interparietalis, is a dense, fibrous connective tissue joint between the two parietal bones of the skull. The term is derived from the Latin word sagitta, meaning arrow.

Jackson–Weiss syndrome (JWS) is a genetic disorder characterized by foot abnormalities and the premature fusion of certain bones of the skull (craniosynostosis), which prevents further growth of the skull and affects the shape of the head and face. This genetic disorder can also sometimes cause intellectual disability and crossed eyes. It was characterized in 1976.
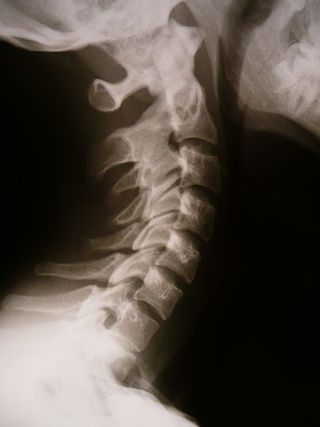
Synostosis is fusion of two or more bones. It can be normal in puberty, fusion of the epiphyseal plate to become the epiphyseal line, or abnormal. When synostosis is abnormal it is a type of dysostosis. Examples of synostoses include:
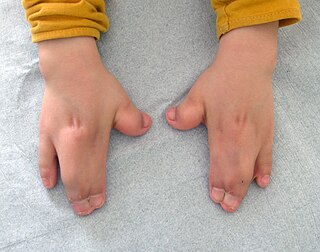
Acrocephalosyndactyly is a group of congenital conditions characterized by irregular features of the face and skull (craniosynostosis) and hands and feet (syndactyly). Craniosynostosis occurs when the cranial sutures, the fibrous tissue connecting the skull bones, fuse the cranial bones early in development. Cranial sutures allow the skull bones to continue growing until they fuse at age 24. Premature fusing of the cranial sutures can result in alterations to the skull shape and interfere with brain growth. Syndactyly occurs when digits of the hands or feet are fused together. When polydactyly is also present, the classification is acrocephalopolysyndactyly. Polydactyly occurs when the hands or feet possess additional digits. Acrocephalosyndactyly is usually diagnosed after birth, although prenatal diagnosis is sometimes possible if the genetic variation is present in family members, as the conditions are typically inherited in an autosomal dominant pattern Treatment often involves surgery in early childhood to correct for craniosynostosis and syndactyly.
Muenke syndrome, also known as FGFR3-related craniosynostosis, is a human specific condition characterized by the premature closure of certain bones of the skull during development, which affects the shape of the head and face. First described by Maximilian Muenke, the syndrome occurs in about 1 in 30,000 newborns. This condition accounts for an estimated 8 percent of all cases of craniosynostosis.

The calvaria is the top part of the skull. It is the superior part of the neurocranium and covers the cranial cavity containing the brain. It forms the main component of the skull roof.

Baller–Gerold syndrome (BGS) is a rare genetic syndrome that involves premature fusion of the skull bones and malformations of facial, forearm and hand bones. The symptoms of Baller–Gerold syndrome overlap with features of a few other genetics disorders: Rothmund–Thomson syndrome and RAPADILINO syndrome. The prevalence of BGS is unknown, as there have only been a few reported cases, but it is estimated to be less than 1 in a million. The name of the syndrome comes from the researchers Baller and Gerold who discovered the first three cases.

Kleeblattschaedel is a rare malformation of the head where there is a protrusion of the skull and broadening of the face. This condition is a severe type of craniosynostosis.
