Perforin-1 (PRF) is a pore-forming protein encoded in humans by the PRF1gene. It is stored in the secretory granules of cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells, collectively known as cytotoxic lymphocytes (CLs). Upon activation, these cells release perforin to form pores in the membranes of target cells, enabling the entry of granzymes that trigger apoptosis. Perforin is therefore a central effector molecule of the immune system, essential for the elimination of virus-infected and transformed cells.[5] Mutations in PRF1 that impair perforin expression or function are associated with familial hemophagocytic lymphohistiocytosis (FHL) and related immune dysregulation syndromes, a spectrum of conditions sometimes collectively referred to as perforinopathies.[5]
Perforin was initially discovered in 1983 and subsequently cloned from an expression library in 1988 using anti-complement C9 antibody cross-reactivity. A sequence comparison showed a notable resemblance between the two proteins in a specific central region, termed the 'membrane attack complex/perforin' (MACPF) domain.[6]
Structure
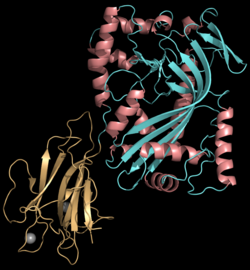
Perforin is a pore-forming cytolytic protein composed of approximately 555 amino acids and has a molecular weight of 60–70 kDa. The protein contains several domains: the conserved N-terminal membrane attack complex/perforin (MACPF) domain which is central to its pore-forming function, a C-terminal membrane-docking C2 domain responsible for calcium-dependent interaction with target membranes, and an epidermal growth factor (EGF)-like domain that provides flexibility and links the MACPF and C2 domains. The structure of perforin is further stabilized by nine disulfide bonds, and its N-terminal domain binds calcium ions, a key feature required for activation and subsequent insertion into lipid membranes. Oligomerization of approximately 20 perforin monomers forms large, cylindrical pores in target cell membranes; these pores are hydrophobic and disrupt ionic homeostasis, leading to cell death.[7][8]
The lytic membrane-inserting region of perforin is the MACPF domain, which mediates pore formation.[9] This domain shares homology with cholesterol-dependent cytolysins of Gram-positive bacteria.[10] Perforin also shows structural similarity to complement component 9 (C9), another pore-forming protein that creates transmembrane tubules.[11]
Purifying perforin has historically been difficult due to its loss of activity and stability in solution; only recently has a recombinant form been successfully produced.[12]
Function
Perforin is a pore-forming cytolytic protein stored in the granules of cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells. Upon degranulation, perforin is escorted to the target cell membrane by calreticulin, a chaperone protein that prevents its premature degradation. Perforin binds to the target cell's plasma membrane through interactions with membrane phospholipids, while calcium ions enhance this binding by stabilizing interactions with phosphatidylcholine[7] In a Ca2+-dependent process, perforin oligomerises to form pores that permit the entry of granzymes, a family of pro-apoptotic proteases.[13]
Initially, perforin was thought to act only at the plasma membrane. However, subsequent findings revealed that granzyme B can be endocytosed independently of perforin. Washed cells that had internalized granzyme B underwent apoptosis when perforin was later added, even though perforin had not been present during endocytosis. These results led to the proposal that perforin's main function occurs at the endosomal rather than the plasma membrane, by disrupting endosomal integrity to release granzymes into the cytosol.[14][12] Later studies confirmed that perforin pores in the endosomal membrane enable granzyme B to escape into the cytosol, thereby triggering apoptosis.[15]
Through these mechanisms, perforin acts as a central effector molecule in CTL- and NK cell-mediated cytotoxicity.
Clinical significance
Familial hemophagocytic lymphohistiocytosis
Mutations in PRF1 that reduce or abolish perforin expression or pore-forming activity cause the autosomal-recessive disorder familial hemophagocytic lymphohistiocytosis (FHL) type 2 (FHL2). The loss of cytotoxic T lymphocyte (CTL) and natural killer (NK) cell function prevents effective granule-mediated cytotoxicity, leading to uncontrolled antigen presentation, T-cell hyperactivation, interferon-γ–driven macrophage activation, and severe hyperinflammation. PRF1 mutations account for roughly 20–50% of familial cases, with disease severity depending on mutation type: hypomorphic alleles with residual activity may present later in childhood or adulthood, whereas null mutations typically manifest in infancy.[16][5][17][18]
Perforinopathy
The concept of "perforinopathy" encompasses a spectrum of disease presentations linked to impaired perforin function.[5]
Acute
Complete loss of perforin activity causes a severe, often fatal, autosomal recessive immunoregulatory disorder in infants, typically presenting before 12 months of age as FHL. Effective treatment requires allogeneic bone marrow transplantation.[5] Pathogenesis results from the inability of CTLs and NK cells to kill target cells, leading to the clinical syndrome defined in HLH-2004. Diagnosis is confirmed by impaired NK cell cytotoxicity and by mutations in PRF1 or other FHL-associated genes such as UNC13D, STX11, and STXBP2.[5]
Sub-acute
Sub-acute perforinopathies result from partial loss of CTL and NK cell function, usually due to bi-allelic hypomorphic mutations. Clinical manifestations are more variable and often milder than in acute disease, with intermittent courses, later onset, and responsiveness to immunosuppressive or immune-ablative therapy. These features make diagnosis more challenging.[5]
Chronic
Chronic perforinopathies arise from monoallelic mutations in genes linked to FHL. Instead of classic FHL, these patients may present with later-onset immune dysregulation, including macrophage activation syndrome in juvenile rheumatoid arthritis or an increased incidence of blood cancers. Symptoms usually appear after age 5. Associations between PRF1 variants and outcomes following bone marrow transplantation have been reported, but remain controversial.[5]
Cancer
Perforin is a central effector in immune surveillance against cancer, enabling CTLs and NK cells to lyse transformed cells (a normal cell altered to grow uncontrollably with cancer-like traits). By forming membrane pores, perforin permits entry of granzymes that induce apoptosis. In both humans and mice, perforin deficiency or dysfunction markedly increases susceptibility to cancers, particularly lymphomas and other hematological malignancies. Some tumors evade perforin-mediated cytotoxicity by altering cell surface molecules, thereby resisting immune clearance and promoting cancer progression.[19][20][7][21]
↑ Trapani JA (December 1995). "Target cell apoptosis induced by cytotoxic T cells and natural killer cells involves synergy between the pore-forming protein, perforin, and the serine protease, granzyme B". Australian and New Zealand Journal of Medicine. 25 (6): 793–799. doi:10.1111/j.1445-5994.1995.tb02883.x. PMID8770355.
↑ Cullen SP, Brunet M, Martin SJ (April 2010). "Granzymes in cancer and immunity". Cell Death and Differentiation. 17 (4): 616–23. doi:10.1038/cdd.2009.206. PMID20075940.
↑ Andrin C, Pinkoski MJ, Burns K, Atkinson EA, Krahenbuhl O, Hudig D, etal. (July 1998). "Interaction between a Ca2+-binding protein calreticulin and perforin, a component of the cytotoxic T-cell granules". Biochemistry. 37 (29): 10386–10394. doi:10.1021/bi980595z. PMID9671507.
Further reading
Trapani JA (December 1995). "Target cell apoptosis induced by cytotoxic T cells and natural killer cells involves synergy between the pore-forming protein, perforin, and the serine protease, granzyme B". Australian and New Zealand Journal of Medicine. 25 (6): 793–799. doi:10.1111/j.1445-5994.1995.tb02883.x. PMID8770355.
Peitsch MC, Amiguet P, Guy R, Brunner J, Maizel JV, Tschopp J (July 1990). "Localization and molecular modelling of the membrane-inserted domain of the ninth component of human complement and perforin". Molecular Immunology. 27 (7): 589–602. doi:10.1016/0161-5890(90)90001-G. PMID2395434.
Young JD, Hengartner H, Podack ER, Cohn ZA (March 1986). "Purification and characterization of a cytolytic pore-forming protein from granules of cloned lymphocytes with natural killer activity". Cell. 44 (6): 849–859. doi:10.1016/0092-8674(86)90007-3. PMID2420467. S2CID30182487.
Young JD, Cohn ZA, Podack ER (July 1986). "The ninth component of complement and the pore-forming protein (perforin 1) from cytotoxic T cells: structural, immunological, and functional similarities". Science. 233 (4760): 184–190. doi:10.1126/science.2425429. PMID2425429.
Goebel WS, Schloemer RH, Brahmi Z (1996). "Target cell-induced perforin mRNA turnover in NK3.3 cells is mediated by multiple elements within the mRNA coding region". Molecular Immunology. 33 (4–5): 341–349. doi:10.1016/0161-5890(95)00155-7. PMID8676885.
Andrin C, Pinkoski MJ, Burns K, Atkinson EA, Krahenbuhl O, Hudig D, etal. (July 1998). "Interaction between a Ca2+-binding protein calreticulin and perforin, a component of the cytotoxic T-cell granules". Biochemistry. 37 (29): 10386–10394. doi:10.1021/bi980595z. PMID9671507.
Ambach A, Bonnekoh B, Gollnick H (2001). "Perforin granule release from cytotoxic lymphocytes ex vivo is inhibited by ciclosporin but not by methotrexate". Skin Pharmacology and Applied Skin Physiology. 14 (5): 249–260. doi:10.1159/000056355. PMID11586066. S2CID30142804.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.