Thrombotic thrombocytopenic purpura (TTP) is a blood disorder that results in blood clots forming in small blood vessels throughout the body. This results in a low platelet count, low red blood cells due to their breakdown, and often kidney, heart, and brain dysfunction. Symptoms may include large bruises, fever, weakness, shortness of breath, confusion, and headache. Repeated episodes may occur.

Pyruvate kinase is the enzyme involved in the last step of glycolysis. It catalyzes the transfer of a phosphate group from phosphoenolpyruvate (PEP) to adenosine diphosphate (ADP), yielding one molecule of pyruvate and one molecule of ATP. Pyruvate kinase was inappropriately named before it was recognized that it did not directly catalyze phosphorylation of pyruvate, which does not occur under physiological conditions. Pyruvate kinase is present in four distinct, tissue-specific isozymes in animals, each consisting of particular kinetic properties necessary to accommodate the variations in metabolic requirements of diverse tissues.

Phosphofructokinase deficiency is a rare muscular metabolic disorder, with an autosomal recessive inheritance pattern. It is characterized as a deficiency in the Phosphofructokinase (PFK) enzyme throughout the body, including the skeletal muscles and red blood cells. Phosphofrucotkinase is an enzyme involved in the glycolytic process. The lack of PFK blocks the completion of the glycolytic pathway. Therefore, all products past the block would be deficient, including Adenosine triphosphate (ATP).

Pyruvate kinase deficiency is an inherited metabolic disorder of the enzyme pyruvate kinase which affects the survival of red blood cells. Both autosomal dominant and recessive inheritance have been observed with the disorder; classically, and more commonly, the inheritance is autosomal recessive. Pyruvate kinase deficiency is the second most common cause of enzyme-deficient hemolytic anemia, following G6PD deficiency.
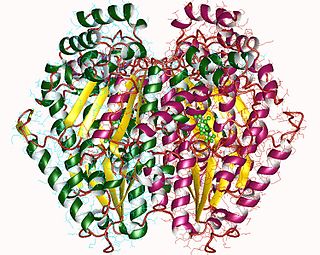
Glucose-6-phosphate isomerase (GPI), alternatively known as phosphoglucose isomerase/phosphoglucoisomerase (PGI) or phosphohexose isomerase (PHI), is an enzyme that in humans is encoded by the GPI gene on chromosome 19. This gene encodes a member of the glucose phosphate isomerase protein family. The encoded protein has been identified as a moonlighting protein based on its ability to perform mechanistically distinct functions. In the cytoplasm, the gene product functions as a glycolytic enzyme that interconverts glucose-6-phosphate (G6P) and fructose-6-phosphate (F6P). Extracellularly, the encoded protein functions as a neurotrophic factor that promotes survival of skeletal motor neurons and sensory neurons, and as a lymphokine that induces immunoglobulin secretion. The encoded protein is also referred to as autocrine motility factor (AMF) based on an additional function as a tumor-secreted cytokine and angiogenic factor. Defects in this gene are the cause of nonspherocytic hemolytic anemia, and a severe enzyme deficiency can be associated with hydrops fetalis, immediate neonatal death and neurological impairment. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Jan 2014]

Hereditary stomatocytosis describes a number of inherited, mostly autosomal dominant human conditions which affect the red blood cell and create the appearance of a slit-like area of central pallor (stomatocyte) among erythrocytes on peripheral blood smear. The erythrocytes' cell membranes may abnormally 'leak' sodium and/or potassium ions, causing abnormalities in cell volume. Hereditary stomatocytosis should be distinguished from acquired causes of stomatocytosis, including dilantin toxicity and alcoholism, as well as artifact from the process of preparing peripheral blood smears.

Erythrocyte membrane protein band 4.2 is a protein that in humans is encoded by the EPB42 gene. It is part of the red blood cell cytoskeleton.

Aldolase A deficiency is an autosomal recessive metabolic disorder resulting in a deficiency of the enzyme aldolase A; the enzyme is found predominantly in red blood cells and muscle tissue. The deficiency may lead to hemolytic anaemia as well as myopathy associated with exercise intolerance and rhabdomyolysis in some cases.

Phosphoglycerate kinase 1 is an enzyme that in humans is encoded by the PGK1 gene.
Hexokinase-1 (HK1) is an enzyme that in humans is encoded by the HK1 gene on chromosome 10. Hexokinases phosphorylate glucose to produce glucose-6-phosphate (G6P), the first step in most glucose metabolism pathways. This gene encodes a ubiquitous form of hexokinase which localizes to the outer membrane of mitochondria. Mutations in this gene have been associated with hemolytic anemia due to hexokinase deficiency. Alternative splicing of this gene results in five transcript variants which encode different isoforms, some of which are tissue-specific. Each isoform has a distinct N-terminus; the remainder of the protein is identical among all the isoforms. A sixth transcript variant has been described, but due to the presence of several stop codons, it is not thought to encode a protein. [provided by RefSeq, Apr 2009]

Spectrin alpha chain, erythrocyte is a protein that in humans is encoded by the SPTA1 gene.

Glutamate–cysteine ligase catalytic subunit is an enzyme that in humans is encoded by the GCLC gene.

Cytosolic 5'-nucleotidase 3 (NTC53), also known as cytosolic 5'-nucleotidase 3A, pyrimidine 5’-nucleotidase, and p56, is an enzyme that in humans is encoded by the NT5C3, or NT5C3A, gene on chromosome 7.

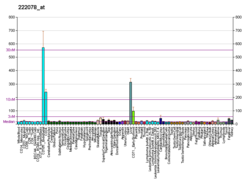
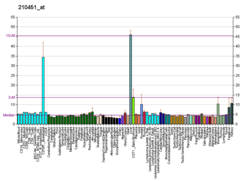
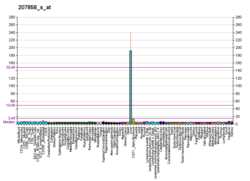
Pyruvate kinase isozymes M1/M2 (PKM1/M2), also known as pyruvate kinase muscle isozyme (PKM), pyruvate kinase type K, cytosolic thyroid hormone-binding protein (CTHBP), thyroid hormone-binding protein 1 (THBP1), or opa-interacting protein 3 (OIP3), is an enzyme that in humans is encoded by the PKM2 gene.

Hexokinase deficiency is an extremely rare autosomal recessive condition that falls under the category of erythroenzymopathies, or defects in red cell enzymes. Hexokinase deficiency manifests is associated with chronic nonspherocytic hemolytic anemia. Hemolytic anemia seems to be the only clinical sign of hexokinase deficiency. In 1967 the first case of hexokinase deficiency was described by Valentine et al, since then, less than 50 cases have been reported.
Congenital hemolytic anemia (CHA) is a diverse group of rare hereditary conditions marked by decreased life expectancy and premature removal of erythrocytes from blood flow. Defects in erythrocyte membrane proteins and red cell enzyme metabolism, as well as changes at the level of erythrocyte precursors, lead to impaired bone marrow erythropoiesis. CHA is distinguished by variable anemia, chronic extravascular hemolysis, decreased erythrocyte life span, splenomegaly, jaundice, biliary lithiasis, and iron overload. Immune-mediated mechanisms may play a role in the pathogenesis of these uncommon diseases, despite the paucity of data regarding the immune system's involvement in CHAs.

Glutaredoxin 5, also known as GLRX5, is a protein which in humans is encoded by the GLRX5 gene located on chromosome 14. This gene encodes a mitochondrial protein, which is evolutionarily conserved. It is involved in the biogenesis of iron- sulfur clusters, which are required for normal iron homeostasis. Mutations in this gene are associated with autosomal recessive pyridoxine-refractory sideroblastic anemia.
Human genetic resistance to malaria refers to inherited changes in the DNA of humans which increase resistance to malaria and result in increased survival of individuals with those genetic changes. The existence of these genotypes is likely due to evolutionary pressure exerted by parasites of the genus Plasmodium which cause malaria. Since malaria infects red blood cells, these genetic changes are most common alterations to molecules essential for red blood cell function, such as hemoglobin or other cellular proteins or enzymes of red blood cells. These alterations generally protect red blood cells from invasion by Plasmodium parasites or replication of parasites within the red blood cell.

Adenylate kinase 1 is a protein that in humans is encoded by the AK1 gene.

Mitapivat, sold under the brand name Pyrukynd, is a medication used to treat hemolytic anemia. It is taken as the sulfate hydrate salt by mouth. Mitapivat is a pyruvate kinase activator.