
The aldol reaction is a reaction in organic chemistry that combines two carbonyl compounds to form a new β-hydroxy carbonyl compound. Its simplest form might involve the nucleophilic addition of an enolized ketone to another:
In organic chemistry, a nucleophilic addition reaction is an addition reaction where a chemical compound with an electrophilic double or triple bond reacts with a nucleophile, such that the double or triple bond is broken. Nucleophilic additions differ from electrophilic additions in that the former reactions involve the group to which atoms are added accepting electron pairs, whereas the latter reactions involve the group donating electron pairs.

In organic chemistry, the Michael reaction or Michael 1,4 addition is a reaction between a Michael donor and a Michael acceptor to produce a Michael adduct by creating a carbon-carbon bond at the acceptor's β-carbon. It belongs to the larger class of conjugate additions and is widely used for the mild formation of carbon-carbon bonds.
The Hiyama coupling is a palladium-catalyzed cross-coupling reaction of organosilanes with organic halides used in organic chemistry to form carbon–carbon bonds. This reaction was discovered in 1988 by Tamejiro Hiyama and Yasuo Hatanaka as a method to form carbon-carbon bonds synthetically with chemo- and regioselectivity. The Hiyama coupling has been applied to the synthesis of various natural products.
The aza-Baylis–Hillman reaction or aza-BH reaction in organic chemistry is a variation of the Baylis–Hillman reaction and describes the reaction of an electron deficient alkene, usually an α,β-unsaturated carbonyl compound, with an imine in the presence of a nucleophile. The reaction product is an allylic amine. The reaction can be carried out in enantiomeric excess of up to 90% with the aid of bifunctional chiral BINOL and phosphinyl BINOL compounds, for example in the reaction of n-(4-chloro-benzylidene)-benzenesulfonamide with methyl vinyl ketone (MVK) in cyclopentyl methyl ether and toluene at -15°C.
In organic chemistry, umpolung or polarity inversion is the chemical modification of a functional group with the aim of the reversal of polarity of that group. This modification allows secondary reactions of this functional group that would otherwise not be possible. The concept was introduced by D. Seebach and E.J. Corey. Polarity analysis during retrosynthetic analysis tells a chemist when umpolung tactics are required to synthesize a target molecule.
In organosilicon chemistry, silyl enol ethers are a class of organic compounds that share the common functional group R3Si−O−CR=CR2, composed of an enolate bonded to a silane through its oxygen end and an ethene group as its carbon end. They are important intermediates in organic synthesis.

In organic chemistry, organocatalysis is a form of catalysis in which the rate of a chemical reaction is increased by an organic catalyst. This "organocatalyst" consists of carbon, hydrogen, sulfur and other nonmetal elements found in organic compounds. Because of their similarity in composition and description, they are often mistaken as a misnomer for enzymes due to their comparable effects on reaction rates and forms of catalysis involved.

In organic chemistry, the Mukaiyama aldol addition is an organic reaction and a type of aldol reaction between a silyl enol ether and an aldehyde or formate. The reaction was discovered by Teruaki Mukaiyama in 1973. His choice of reactants allows for a crossed aldol reaction between an aldehyde and a ketone, or a different aldehyde without self-condensation of the aldehyde. For this reason the reaction is used extensively in organic synthesis.
Chiral Lewis acids (CLAs) are a type of Lewis acid catalyst. These acids affect the chirality of the substrate as they react with it. In such reactions, synthesis favors the formation of a specific enantiomer or diastereomer. The method is an enantioselective asymmetric synthesis reaction. Since they affect chirality, they produce optically active products from optically inactive or mixed starting materials. This type of preferential formation of one enantiomer or diastereomer over the other is formally known as asymmetric induction. In this kind of Lewis acid, the electron-accepting atom is typically a metal, such as indium, zinc, lithium, aluminium, titanium, or boron. The chiral-altering ligands employed for synthesizing these acids often have multiple Lewis basic sites that allow the formation of a ring structure involving the metal atom.
Within the area of organocatalysis, (thio)urea organocatalysis describes the use of ureas and thioureas to accelerate and stereochemically alter organic transformations. The effects arise through hydrogen-bonding interactions between the substrate and the (thio)urea. Unlike classical catalysts, these organocatalysts interact by non-covalent interactions, especially hydrogen bonding. The scope of these small-molecule H-bond donors termed (thio)urea organocatalysis covers both non-stereoselective and stereoselective applications.
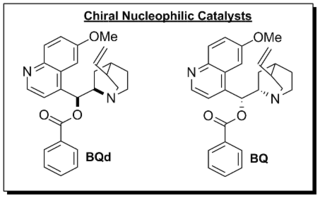
Lectka and co-workers developed a catalytic, asymmetric method to synthesize β-lactams.
Trimethylenemethane cycloaddition is the formal (3+2) annulation of trimethylenemethane (TMM) derivatives to two-atom pi systems. Although TMM itself is too reactive and unstable to be stored, reagents which can generate TMM or TMM synthons in situ can be used to effect cycloaddition reactions with appropriate electron acceptors. Generally, electron-deficient pi bonds undergo cyclization with TMMs more easily than electron-rich pi bonds.
Reductions with hydrosilanes are methods used for hydrogenation and hydrogenolysis of organic compounds. The approach is a subset of ionic hydrogenation. In this particular method, the substrate is treated with a hydrosilane and auxiliary reagent, often a strong acid, resulting in formal transfer of hydride from silicon to carbon. This style of reduction with hydrosilanes enjoys diverse if specialized applications.
In organic chemistry, the Baylis–Hillman, Morita–Baylis–Hillman, or MBH reaction is a carbon-carbon bond-forming reaction between an activated alkene and a carbon electrophile in the presence of a nucleophilic catalyst, such as a tertiary amine or phosphine. The product is densely functionalized, joining the alkene at the α-position to a reduced form of the electrophile.
The Tsuji–Trost reaction is a palladium-catalysed substitution reaction involving a substrate that contains a leaving group in an allylic position. The palladium catalyst first coordinates with the allyl group and then undergoes oxidative addition, forming the π-allyl complex. This allyl complex can then be attacked by a nucleophile, resulting in the substituted product.

Hydrogen-bond catalysis is a type of organocatalysis that relies on use of hydrogen bonding interactions to accelerate and control organic reactions. In biological systems, hydrogen bonding plays a key role in many enzymatic reactions, both in orienting the substrate molecules and lowering barriers to reaction. However, chemists have only recently attempted to harness the power of using hydrogen bonds to perform catalysis, and the field is relatively undeveloped compared to research in Lewis acid catalysis.

Scott Eric Denmark is an American chemist who is the Reynold C. Fuson Professor of Chemistry at the University of Illinois at Urbana-Champaign (UIUC). Denmark received an S.B. degree from MIT in 1975 and the D.Sc.Tech. degree from ETH Zurich in 1980, under the supervision of Professor Albert Eschenmoser. He joined the faculty at UIUC the same year and became an associate professor in 1986, full professor in 1987, and was named the Fuson Professor of Chemistry in 1991. He served as the president and editor-in-chief of the Organic Reactions book series between 2008 and 2018. In 2017, Denmark was elected to the American Academy of Arts and Sciences. In 2018, he was elected to the National Academy of Sciences.

Teruaki Mukaiyama was a Japanese organic chemist. One of the most prolific chemists of the 20th century in the field of organic synthesis, Mukaiyama helped establish the field of organic chemistry in Japan after World War II.
Palladium forms a variety of ionic, coordination, and organopalladium compounds, typically with oxidation state Pd0 or Pd2+. Pd3+ compounds have also been reported. Palladium compounds are frequently used as catalysts in cross-coupling reactions such as the Sonogashira coupling and Suzuki reaction.
