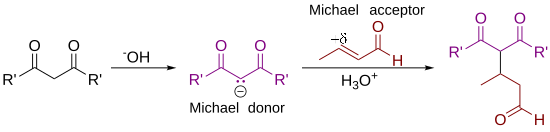
In organic chemistry, the Michael reaction or Michael 1,4 addition is a reaction between a Michael donor (an enolate or other nucleophile) and a Michael acceptor (usually an α,β-unsaturated carbonyl) to produce a Michael adduct by creating a carbon-carbon bond at the acceptor's β-carbon.[1][2] It belongs to the larger class of conjugate additions and is widely used for the mild formation of carbon–carbon bonds.[3]
As originally defined by Arthur Michael,[7][8] the reaction is the addition of an enolate of a ketone or aldehyde to an α,β-unsaturated carbonyl compound at the β carbon. The current definition of the Michael reaction has broadened to include nucleophiles other than enolates.[9] Some examples of nucleophiles include doubly stabilized carbon nucleophiles such as beta-ketoesters, malonates, and beta-cyanoesters. The resulting product contains a highly useful 1,5-dioxygenated pattern. Non-carbon nucleophiles such as water, alcohols, amines, and enamines can also react with an α,β-unsaturated carbonyl in a 1,4-addition.[10]
Some authors have broadened the definition of the Michael addition to essentially refer to any 1,4-addition reaction of α,β-unsaturated carbonyl compounds. Others, however, insist that such a usage is an abuse of terminology, and limit the Michael addition to the formation of carbon–carbon bonds through the addition of carbon nucleophiles. The terms oxa-Michael reaction and aza-Michael reaction[2] have been used to refer to the 1,4-addition of oxygen and nitrogen nucleophiles, respectively. The Michael reaction has also been associated with 1,6-addition reactions.[11]
Deprotonation of 1 by a base leads to carbanion2, stabilized by its electron-withdrawing groups. Structures 2a to 2c are three resonance structures that can be drawn for this species, two of which have enolate ions. This nucleophile reacts with the electrophilic alkene 3 to form 4 in a conjugate addition reaction. Finally, enolate 4 abstracts a proton from protonated base (or solvent) to produce 5.
The reaction is dominated by orbital, rather than electrostatic, considerations. The HOMO of stabilized enolates has a large coefficient on the central carbon atom while the LUMO of many alpha, beta unsaturated carbonyl compounds has a large coefficient on the beta carbon. Thus, both reactants can be considered soft. These polarized frontier orbitals are of similar energy, and react efficiently to form a new carbon–carbon bond.[12]
Like the aldol addition, the Michael reaction may proceed via an enol, silyl enol ether in the Mukaiyama–Michael addition, or more usually, enolate nucleophile. In the latter case, the stabilized carbonyl compound is deprotonated with a strong base (hard enolization) or with a Lewis acid and a weak base (soft enolization). The resulting enolate attacks the activated olefin with 1,4-regioselectivity, forming a carbon–carbon bond. This also transfers the enolate to the electrophile. Since the electrophile is much less acidic than the nucleophile, rapid proton transfer usually transfers the enolate back to the nucleophile if the product is enolizable; however, one may take advantage of the new locus of nucleophilicity if a suitable electrophile is pendant. Depending on the relative acidities of the nucleophile and product, the reaction may be catalytic in base. In most cases, the reaction is irreversible at low temperature.
History
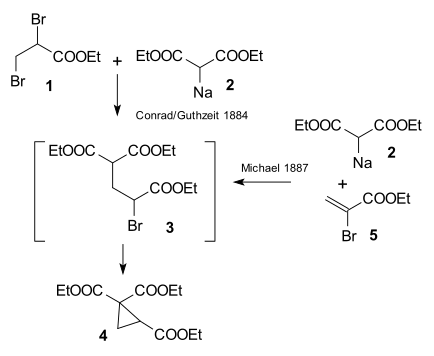
The research done by Arthur Michael in 1887 at Tufts University was prompted by an 1884 publication by Conrad & Kuthzeit on the reaction of ethyl 2,3-dibromopropionate with diethyl sodiomalonate forming a cyclopropane derivative[13] (now recognized as involving two successive substitution reactions).
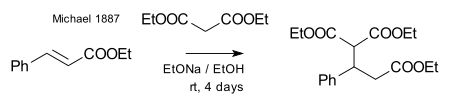
Michael was able to obtain the same product by replacing the propionate by 2-bromacrylic acid ethylester and realized that this reaction could only work by assuming an addition reaction to the double bond of the acrylic acid. He then confirmed this assumption by reacting diethyl malonate and the ethyl ester of cinnamic acid forming the first Michael adduct:[14]
In the same year Rainer Ludwig Claisen claimed priority for the invention.[15] He and T. Komnenos had observed addition products to double bonds as side-products earlier in 1883 while investigating condensation reactions of malonic acid with aldehydes.[16] However, according to biographer Takashi Tokoroyama, this claim is without merit.[14]
Syn addition is favored with 99% ee. In the transition state believed to be responsible for this selectivity, the enamine (formed between the proline nitrogen and the cycloketone) and β-nitrostyrene are co-facial with the nitro group hydrogen bonded to the protonated amine in the proline side group.
The 1,6-Michael reaction proceeds via nucleophilic attack on the 𝛿 carbon of an α,β-,𝛿-diunsaturated Michael acceptor.[34][35] The 1,6-addition mechanism is similar to the 1,4-addition, with one exception being the nucleophilic attack occurring at the 𝛿 carbon of the Michael acceptor.[35] However, research shows that organocatalysis often favours the 1,4-addition.[34] In many syntheses where 1,6-addition was favoured, the substrate contained certain structural features.[35] Research has shown that catalysts can also influence the regioselectivity and enantioselectivity of a 1,6-addition reaction.[35]
For example, the image below shows the addition of ethylmagnesium bromide to ethyl sorbate 1 using a copper catalyst with a reversed josiphos (R,S)-(–)-3 ligand.[35] This reaction produced the 1,6-addition product 2 in 0% yield, the 1,6-addition product 3 in approximately 99% yield, and the 1,4-addition product 4 in less than 2% yield. This particular catalyst and set of reaction conditions led to the mostly regioselective and enantioselective 1,6-Michael addition of ethyl sorbate 1 to product 3.
The Michael addition of ethylmagnesium bromide to ethyl sorbate.
Applications
Pharmaceuticals
A Michael reaction is used as a mechanistic step by many covalent inhibitor drugs. Cancer drugs such as ibrutinib, osimertinib, and rociletinib have an acrylamide functional group as a Michael acceptor. The Michael donor on the drug reacts with a Michael acceptor in the active site of an enzyme. This is a viable cancer treatment because the target enzyme is inhibited following the Michael reaction.[36]
All polymerization reactions have three basic steps: initiation, propagation, and termination. The initiation step is the Michael addition of the nucleophile to a monomer. The resultant species undergoes a Michael addition with another monomer, with the latter acting as an acceptor. This extends the chain by forming another nucleophilic species to act as a donor for the next addition. This process repeats until the reaction is quenched by chain termination.[37] The original Michael donor can be a neutral donor such as amines, thiols, and alkoxides, or alkyl ligands bound to a metal.[38]
Polymerization mechanism of a Michael addition with a thiol nucleophile
Examples
Linear step growth polymerizations are some of the earliest applications of the Michael reaction in polymerizations. A wide variety of Michael donors and acceptors have been used to synthesize a diverse range of polymers. Examples of such polymers include poly(amido amine), poly(amino ester), poly(imido sulfide), poly(ester sulfide), poly(aspartamide), poly(imido ether), poly(amino quinone), poly(enone sulfide) and poly(enamine ketone).
For example, linear step growth polymerization produces the redox active poly(amino quinone), which serves as an anti-corrosion coatings on various metal surfaces.[39] Another example includes network polymers, which are used for drug delivery, high performance composites, and coatings. These network polymers are synthesized using a dual chain growth, photo-induced radical and step growth Michael addition system.
↑ Brown, William Henry (2018). Organic chemistry. Brent L. Iverson, Eric V. Anslyn, Christopher S. Foote (Eighthed.). Boston, Mass. ISBN978-1-337-51640-2. OCLC1200494733.{{cite book}}: CS1 maint: location missing publisher (link)
1 2 Tokoroyama, T. (2010). "Discovery of the Michael Reaction". European Journal of Organic Chemistry. 2010 (10): 2009–2016. doi:10.1002/ejoc.200901130.
↑ Pansare, S. V.; Pandya, K. (2006). "Simple Diamine- and Triamine-Protonic Acid Catalysts for the Enantioselective Michael Addition of Cyclic Ketones to Nitroalkenes". Journal of the American Chemical Society. 128 (30): 9624–9625. Bibcode:2006JAChS.128.9624P. doi:10.1021/ja062701n. PMID16866504.
↑ Ikawa, M.; Stahmann, M. A.; Link, K. P. (1944). "Studies on 4-Hydroxycoumarins. V. The Condensation of α,β-Unsaturated Ketones with 4-Hydroxycoumarin". Journal of the American Chemical Society. 66 (6): 902. Bibcode:1944JAChS..66..902I. doi:10.1021/ja01234a019.
↑ Halland, N.; Hansen, T.; Jørgensen, K. (2003). "Organocatalytic asymmetric Michael reaction of cyclic 1,3-dicarbonyl compounds and α,β-unsaturated ketones--a highly atom-economic catalytic one-step formation of optically active warfarin anticoagulant". Angewandte Chemie. 42 (40): 4955–4957. doi:10.1002/anie.200352136. PMID14579449.
↑ Kim, H.; Yen, C.; Preston, P.; Chin, J. (2006). "Substrate-directed stereoselectivity in vicinal diamine-catalyzed synthesis of warfarin". Organic Letters. 8 (23): 5239–5242. doi:10.1021/ol062000v. PMID17078687.
↑ Xie, J.; Yue, L.; Chen, W.; Du, W.; Zhu, J.; Deng, J.; Chen, Y. (2007). "Highly Enantioselective Michael Addition of Cyclic 1,3-Dicarbonyl Compounds to α,β-Unsaturated Ketones". Organic Letters. 9 (3): 413–415. doi:10.1021/ol062718a. PMID17249775.
↑ Dong, Z.; Wang, L.; Chen, X.; Liu, X.; Lin, L.; Feng, X. (2009). "Organocatalytic Enantioselective Michael Addition of 4-Hydroxycoumarin to α,β-Unsaturated Ketones: A Simple Synthesis of Warfarin". European Journal of Organic Chemistry. 2009 (30): 5192. doi:10.1002/ejoc.200900831.
↑ Wong, T. C.; Sultana, C. M.; Vosburg, D. A. (2010). "A Green, Enantioselective Synthesis of Warfarin for the Undergraduate Organic Laboratory". Journal of Chemical Education. 87 (2): 194. Bibcode:2010JChEd..87..194W. doi:10.1021/ed800040m.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.