
A generegulatory network (GRN) is a collection of molecular regulators that interact with each other and with other substances in the cell to govern the gene expression levels of mRNA and proteins which, in turn, determine the function of the cell. GRN also play a central role in morphogenesis, the creation of body structures, which in turn is central to evolutionary developmental biology (evo-devo).

Computer simulation is the running of a mathematical model on a computer, the model being designed to represent the behaviour of, or the outcome of, a real-world or physical system. The reliability of some mathematical models can be determined by comparing their results to the real-world outcomes they aim to predict. Computer simulations have become a useful tool for the mathematical modeling of many natural systems in physics, astrophysics, climatology, chemistry, biology and manufacturing, as well as human systems in economics, psychology, social science, health care and engineering. Simulation of a system is represented as the running of the system's model. It can be used to explore and gain new insights into new technology and to estimate the performance of systems too complex for analytical solutions.

Mathematical and theoretical biology, or biomathematics, is a branch of biology which employs theoretical analysis, mathematical models and abstractions of living organisms to investigate the principles that govern the structure, development and behavior of the systems, as opposed to experimental biology which deals with the conduction of experiments to test scientific theories. The field is sometimes called mathematical biology or biomathematics to stress the mathematical side, or theoretical biology to stress the biological side. Theoretical biology focuses more on the development of theoretical principles for biology while mathematical biology focuses on the use of mathematical tools to study biological systems, even though the two terms are sometimes interchanged.

CellML is an XML based markup language for describing mathematical models. Although it could theoretically describe any mathematical model, it was originally created with the Physiome Project in mind, and hence used primarily to describe models relevant to the field of biology. This is reflected in its name CellML, although this is simply a name, not an abbreviation. CellML is growing in popularity as a portable description format for computational models, and groups throughout the world are using CellML for modelling or developing software tools based on CellML. CellML is similar to Systems Biology Markup Language SBML but provides greater scope for model modularity and reuse, and is not specific to descriptions of biochemistry.
Modelling biological systems is a significant task of systems biology and mathematical biology. Computational systems biology aims to develop and use efficient algorithms, data structures, visualization and communication tools with the goal of computer modelling of biological systems. It involves the use of computer simulations of biological systems, including cellular subsystems, to both analyze and visualize the complex connections of these cellular processes.
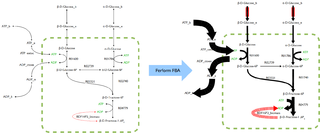
Flux balance analysis (FBA) is a mathematical method for simulating metabolism in genome-scale reconstructions of metabolic networks. In comparison to traditional methods of modeling, FBA is less intensive in terms of the input data required for constructing the model. Simulations performed using FBA are computationally inexpensive and can calculate steady-state metabolic fluxes for large models in a few seconds on modern personal computers. The related method of metabolic pathway analysis seeks to find and list all possible pathways between metabolites.
The Systems Biology Markup Language (SBML) is a representation format, based on XML, for communicating and storing computational models of biological processes. It is a free and open standard with widespread software support and a community of users and developers. SBML can represent many different classes of biological phenomena, including metabolic networks, cell signaling pathways, regulatory networks, infectious diseases, and many others. It has been proposed as a standard for representing computational models in systems biology today.
A stochastic simulation is a simulation of a system that has variables that can change stochastically (randomly) with individual probabilities.
Systems immunology is a research field under systems biology that uses mathematical approaches and computational methods to examine the interactions within cellular and molecular networks of the immune system. The immune system has been thoroughly analyzed as regards to its components and function by using a "reductionist" approach, but its overall function can't be easily predicted by studying the characteristics of its isolated components because they strongly rely on the interactions among these numerous constituents. It focuses on in silico experiments rather than in vivo.
A multi-compartment model is a type of mathematical model used for describing the way materials or energies are transmitted among the compartments of a system. Sometimes, the physical system that we try to model in equations is too complex, so it is much easier to discretize the problem and reduce the number of parameters. Each compartment is assumed to be a homogeneous entity within which the entities being modeled are equivalent. A multi-compartment model is classified as a lumped parameters model. Similar to more general mathematical models, multi-compartment models can treat variables as continuous, such as a differential equation, or as discrete, such as a Markov chain. Depending on the system being modeled, they can be treated as stochastic or deterministic.
Igor I. Goryanin is a systems biologist, who holds a Henrik Kacser Chair in Computational Systems Biology at the University of Edinburgh. He also heads the Biological Systems Unit at the Okinawa Institute of Science and Technology, Japan.

A cellular model is a mathematical model of aspects of a biological cell, for the purposes of in silico research.

Ecolego is a simulation software tool that is used for creating dynamic models and performing deterministic and probabilistic simulations. It is also used for conducting risk assessments of complex dynamic systems evolving over time.
COPASI is an open-source software application for creating and solving mathematical models of biological processes such as metabolic networks, cell-signaling pathways, regulatory networks, infectious diseases, and many others.
Within bioinformatics, intrinsic Noise Analyzer (iNA) is an open source software for studying reaction kinetics in living cells. The software analyzes mathematical models of intracellular reaction kinetics such as gene expression, regulatory networks or signaling pathways to quantify concentration fluctuations due to the random nature of chemical reactions.
Multi-state modeling of biomolecules refers to a series of techniques used to represent and compute the behaviour of biological molecules or complexes that can adopt a large number of possible functional states.

SAAM II, short for "Simulation Analysis and Modeling" version 2.0, is a renowned computer program designed for scientific research in the field of bioscience. It is a descriptive and exploratory tool in drug development, tracers, metabolic disorders, and pharmacokinetics/pharmacodynamics research. It is grounded in the principles of multi-compartment model theory, which is a widely-used approach for modeling complex biological systems. SAAM II facilitates the construction and simulation of models, providing researchers with a friendly user interface allowing the quick run and multi-fitting of simple and complex structures and data. SAAM II is used by many Pharma and Pharmacy Schools as a drug development, research, and educational tool.
The biochemical systems equation is a compact equation of nonlinear differential equations for describing a kinetic model for any network of coupled biochemical reactions and transport processes.
libRoadRunner is a C/C++ software library that supports simulation of SBML based models.. It uses LLVM to generate extremely high-performance code and is the fastest SBML-based simulator currently available. Its main purpose is for use as a reusable library that can be hosted by other applications, particularly on large compute clusters for doing parameter optimization where performance is critical. It also has a set of Python bindings that allow it to be easily used from Python as well as a set of bindings for Julia.
