
Medium-chain acyl-CoA dehydrogenase deficiency is a disorder of fatty acid oxidation that impairs the body's ability to break down medium-chain fatty acids into acetyl-CoA. The disorder is characterized by hypoglycemia and sudden death without timely intervention, most often brought on by periods of fasting or vomiting.
Inborn errors of metabolism form a large class of genetic diseases involving congenital disorders of enzyme activities. The majority are due to defects of single genes that code for enzymes that facilitate conversion of various substances (substrates) into others (products). In most of the disorders, problems arise due to accumulation of substances which are toxic or interfere with normal function, or due to the effects of reduced ability to synthesize essential compounds. Inborn errors of metabolism are often referred to as congenital metabolic diseases or inherited metabolic disorders. Another term used to describe these disorders is "enzymopathies". This term was created following the study of biodynamic enzymology, a science based on the study of the enzymes and their products. Finally, inborn errors of metabolism were studied for the first time by British physician Archibald Garrod (1857–1936), in 1908. He is known for work that prefigured the "one gene–one enzyme" hypothesis, based on his studies on the nature and inheritance of alkaptonuria. His seminal text, Inborn Errors of Metabolism, was published in 1923.

Maple syrup urine disease (MSUD) is a rare, inherited metabolic disorder that affects the body's ability to metabolize amino acids due to a deficiency in the activity of the branched-chain alpha-ketoacid dehydrogenase (BCKAD) complex. It particularly affects the metabolism of amino acids—leucine, isoleucine, and valine. With MSUD, the body is not able to properly break down these amino acids, therefore leading to the amino acids to build up in urine and become toxic. The condition gets its name from the distinctive sweet odor of affected infants' urine and earwax due to the buildup of these amino acids.
In biochemistry and metabolism, beta oxidation (also β-oxidation) is the catabolic process by which fatty acid molecules are broken down in the cytosol in prokaryotes and in the mitochondria in eukaryotes to generate acetyl-CoA. Acetyl-CoA enters the citric acid cycle, generating NADH and FADH2, which are electron carriers used in the electron transport chain. It is named as such because the beta carbon of the fatty acid chain undergoes oxidation and is converted to a carbonyl group to start the cycle all over again. Beta-oxidation is primarily facilitated by the mitochondrial trifunctional protein, an enzyme complex associated with the inner mitochondrial membrane, although very long chain fatty acids are oxidized in peroxisomes.

Systemic primary carnitine deficiency (SPCD) is an inborn error of fatty acid transport caused by a defect in the transporter responsible for moving carnitine across the plasma membrane. Carnitine is an important amino acid for fatty acid metabolism. When carnitine cannot be transported into tissues, fatty acid oxidation is impaired, leading to a variety of symptoms such as chronic muscle weakness, cardiomyopathy, hypoglycemia and liver dysfunction. The specific transporter involved with SPCD is OCTN2, coded for by the SLC22A5 gene located on chromosome 5. SPCD is inherited in an autosomal recessive manner, with mutated alleles coming from both parents.
Numerous genetic disorders are caused by errors in fatty acid metabolism. These disorders may be described as fatty oxidation disorders or as a lipid storage disorders, and are any one of several inborn errors of metabolism that result from enzyme defects affecting the ability of the body to oxidize fatty acids in order to produce energy within muscles, liver, and other cell types.
Carnitine palmitoyltransferase II deficiency, sometimes shortened to CPT-II or CPT2, is an autosomal recessively inherited genetic metabolic disorder characterized by an enzymatic defect that prevents long-chain fatty acids from being transported into the mitochondria for utilization as an energy source. The disorder presents in one of three clinical forms: lethal neonatal, severe infantile hepatocardiomuscular and myopathic.

Mitochondrial trifunctional protein deficiency is an autosomal recessive fatty acid oxidation disorder that prevents the body from converting certain fats to energy, particularly during periods without food.

Malonic aciduria or malonyl-CoA decarboxylase deficiency (MCD) is an autosomal-recessive metabolic disorder caused by a genetic mutation that disrupts the activity of Malonyl-CoA decarboxylase. This enzyme breaks down Malonyl-CoA into acetyl-CoA and carbon dioxide.
Short-chain acyl-coenzyme A dehydrogenase deficiency (SCADD) is an autosomal recessive fatty acid oxidation disorder which affects enzymes required to break down a certain group of fats called short chain fatty acids.

3-Methylcrotonyl-CoA carboxylase deficiency also known as 3-Methylcrotonylglycinuria is an inborn error of leucine metabolism and is inherited through an autosomal recessive fashion. 3-Methylcrotonyl-CoA carboxylase deficiency is caused by mutations in the MCCC1 gene, formerly known as MMCA, or the MCCC2 gene, formerly known as MCCB. MCCC1 encodes the a-subunits of 3-methylcrotonyl-CoA carboxylase while MCCC2 encodes the b-subunits. The clinical presentation of 3-Methylcrotonyl-CoA carboxylase deficiency is varied, even within members of the same family.
2-Methylbutyryl-CoA dehydrogenase deficiency is an autosomal recessive metabolic disorder. It causes the body to be unable to process the amino acid isoleucine properly. Initial case reports identified individuals with developmental delay and epilepsy, however most cases identified through newborn screening have been asymptomatic.

2,4 Dienoyl-CoA reductase also known as DECR1 is an enzyme which in humans is encoded by the DECR1 gene which resides on chromosome 8. This enzyme catalyzes the following reactions

Methionine synthase reductase, also known as MSR, is an enzyme that in humans is encoded by the MTRR gene.

Alpha-aminoadipic semialdehyde synthase is an enzyme encoded by the AASS gene in humans and is involved in their major lysine degradation pathway. It is similar to the separate enzymes coded for by the LYS1 and LYS9 genes in yeast, and related to, although not similar in structure, the bifunctional enzyme found in plants. In humans, mutations in the AASS gene, and the corresponding alpha-aminoadipic semialdehyde synthase enzyme are associated with familial hyperlysinemia. This rare disease is inherited in an autosomal recessive pattern and patients often have no clinical symptoms.

Metabolic myopathies are myopathies that result from defects in biochemical metabolism that primarily affect muscle. They are generally genetic defects that interfere with the ability to create energy, causing a low ATP reservoir within the muscle cell.
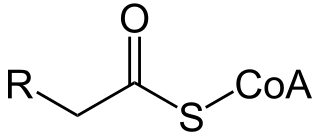
A broad classification for genetic disorders that result from an inability of the body to produce or utilize an enzyme or transport protein that is required to oxidize fatty acids. They are an inborn error of lipid metabolism, and when it affects the muscles also a metabolic myopathy.
Combined malonic and methylmalonic aciduria (CMAMMA), also called combined malonic and methylmalonic acidemia is an inherited metabolic disease characterized by elevated levels of malonic acid and methylmalonic acid. However, the methylmalonic acid levels exceed those of malonic acid. CMAMMA is not only an organic aciduria but also a defect of mitochondrial fatty acid synthesis (mtFASII). Some researchers have hypothesized that CMAMMA might be one of the most common forms of methylmalonic acidemia, and possibly one of the most common inborn errors of metabolism. Due to being infrequently diagnosed, it most often goes undetected.
