Two rules about the percentage of A, C, G, and T in DNA strands
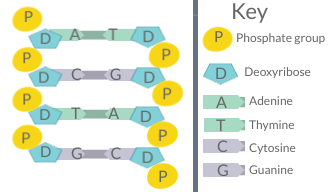
A diagram of DNA base pairing, demonstrating the basis for Chargaff's rules
Chargaff's rules (given by Erwin Chargaff) state that in the DNA of any species and any organism, the amount of guanine should be equal to the amount of cytosine and the amount of adenine should be equal to the amount of thymine. Further, a 1:1 stoichiometric ratio of purine and pyrimidine bases (i.e., A+G=T+C) should exist. This pattern is found in both strands of the DNA. They were discovered by Austrian-born chemist Erwin Chargaff[1][2] in the late 1940s.
The first rule holds that a double-stranded DNA molecule, globally has percentage base pair equality: A% = T% and G% = C%. The rigorous validation of the rule constitutes the basis of Watson–Crick base pairs in the DNA double helix model.
Second parity rule
The second rule holds that both Α% ≈ Τ% and G% ≈ C% are valid for each of the two DNA strands.[3] This describes only a global feature of the base composition in a single DNA strand.[4]
Research
The second parity rule was discovered in 1968.[3] It states that, in single-stranded DNA, the number of adenine units is approximately equal to that of thymine (A% ≈ T%), and the number of cytosine units is approximately equal to that of guanine (C% ≈ G%).
In 2006, it was shown that this rule applies to four[2] of the five types of double stranded genomes; specifically it applies to the eukaryoticchromosomes, the bacterial chromosomes, the double stranded DNA viral genomes, and the archaeal chromosomes.[5] It does not apply to organellar genomes (mitochondria and plastids) smaller than ~20–30 kbp, nor does it apply to single stranded DNA (viral) genomes or any type of RNA genome. The basis for this rule is still under investigation, although genome size may play a role.
Histogram showing how 20309 chromosomes adhere to Chargaff's second parity rule
The rule itself has consequences. In most bacterial genomes (which are generally 80–90% coding) genes are arranged in such a fashion that approximately 50% of the coding sequence lies on either strand. Wacław Szybalski, in the 1960s, showed that in bacteriophage coding sequences purines (A and G) exceed pyrimidines (C and T).[6] This rule has since been confirmed in other organisms and should probably be now termed "Szybalski's rule". While Szybalski's rule generally holds, exceptions are known to exist.[7][8][9] The biological basis for Szybalski's rule is not yet known.
The combined effect of Chargaff's second rule and Szybalski's rule can be seen in bacterial genomes where the coding sequences are not equally distributed. The genetic code has 64 codons of which 3 function as termination codons: there are only 20 amino acids normally present in proteins. (There are two uncommon amino acids—selenocysteine and pyrrolysine—found in a limited number of proteins and encoded by the stop codons—TGA and TAG respectively.) The mismatch between the number of codons and amino acids allows several codons to code for a single amino acid—such codons normally differ only at the third codon base position.
Multivariate statistical analysis of codon use within genomes with unequal quantities of coding sequences on the two strands has shown that codon use in the third position depends on the strand on which the gene is located. This seems likely to be the result of Szybalski's and Chargaff's rules. Because of the asymmetry in pyrimidine and purine use in coding sequences, the strand with the greater coding content will tend to have the greater number of purine bases (Szybalski's rule). Because the number of purine bases will, to a very good approximation, equal the number of their complementary pyrimidines within the same strand and, because the coding sequences occupy 80–90% of the strand, there appears to be (1) a selective pressure on the third base to minimize the number of purine bases in the strand with the greater coding content; and (2) that this pressure is proportional to the mismatch in the length of the coding sequences between the two strands.
Chargaff's 2nd parity rule for prokaryotic 6-mers
The origin of the deviation from Chargaff's rule in the organelles has been suggested to be a consequence of the mechanism of replication.[10] During replication the DNA strands separate. In single stranded DNA, cytosine spontaneously slowly deaminates to adenosine (a C to A transversion). The longer the strands are separated the greater the quantity of deamination. For reasons that are not yet clear the strands tend to exist longer in single form in mitochondria than in chromosomal DNA. This process tends to yield one strand that is enriched in guanine (G) and thymine (T) with its complement enriched in cytosine (C) and adenosine (A), and this process may have given rise to the deviations found in the mitochondria. [citation needed][dubious–discuss]
Chargaff's second rule appears to be the consequence of a more complex parity rule: within a single strand of DNA any oligonucleotide (k-mer or n-gram; length ≤ 10) is present in equal numbers to its reverse complementary nucleotide. Because of the computational requirements this has not been verified in all genomes for all oligonucleotides. It has been verified for triplet oligonucleotides for a large data set.[11] Albrecht-Buehler has suggested that this rule is the consequence of genomes evolving by a process of inversion and transposition.[11] This process does not appear to have acted on the mitochondrial genomes. Chargaff's second parity rule appears to be extended from the nucleotide-level to populations of codon triplets, in the case of whole single-stranded Human genome DNA.[12] A kind of "codon-level second Chargaff's parity rule" is proposed as follows:
Intra-strand relation among percentages of codon populations
First codon
Second codon
Relation proposed
Details
Twx (1st base position is T)
yzA (3rd base position is A)
% Twx% yzA
Twx and yzA are mirror codons, e.g. TCG and CGA
Cwx (1st base position is C)
yzG (3rd base position is G)
% Cwx% yzG
Cwx and yzG are mirror codons, e.g. CTA and TAG
wTx (2nd base position is T)
yAz (2nd base position is A)
% wTx% yAz
wTx and yAz are mirror codons, e.g. CTG and CAG
wCx (2nd base position is C)
yGz (2nd base position is G)
% wCx% yGz
wCx and yGz are mirror codons, e.g. TCT and AGA
wxT (3rd base position is T)
Ayz (1st base position is A)
% wxT% Ayz
wxT and Ayz are mirror codons, e.g. CTT and AAG
wxC (3rd base position is C)
Gyz (1st base position is G)
% wxC% Gyz
wxC and Gyz are mirror codons, e.g. GGC and GCC
Examples — computing whole human genome using the first codons reading frame provides:
36530115 TTT and 36381293 AAA (ratio % = 1.00409). 2087242 TCG and 2085226 CGA (ratio % = 1.00096), etc...
In 2020, it is suggested that the physical properties of the dsDNA (double stranded DNA) and the tendency to maximum entropy of all the physical systems are the cause of Chargaff's second parity rule.[13] The symmetries and patterns present in the dsDNA sequences can emerge from the physical peculiarities of the dsDNA molecule and the maximum entropy principle alone, rather than from biological or environmental evolutionary pressure.
Percentages of bases in DNA
The following table is a representative sample of Erwin Chargaff's 1952 data, listing the base composition of DNA from various organisms and support both of Chargaff's rules.[14] An organism such as φX174 with significant variation from A/T and G/C equal to one, is indicative of single stranded DNA.
↑Szybalski W, Kubinski H, Sheldrick O (1966). "Pyrimidine clusters on the transcribing strand of DNA and their possible role in the initiation of RNA synthesis". Cold Spring Harb Symp Quant Biol. 31: 123–127. doi:10.1101/SQB.1966.031.01.019. PMID4966069.
↑Cristillo AD (1998). Characterization of G0/G1 switch genes in cultured T lymphocytes. Kingston, Ontario, Canada: Queen's University.
↑Nikolaou C, Almirantis Y (2006). "Deviations from Chargaff's second parity rule in organellar DNA. Insights into the evolution of organellar genomes". Gene. 381: 34–41. doi:10.1016/j.gene.2006.06.010. PMID16893615.
Szybalski W, Kubinski H, Sheldrick P (1966). "Pyrimidine clusters on the transcribing strands of DNA and their possible role in the initiation of RNA synthesis". Cold Spring Harbor Symposia on Quantitative Biology. 31: 123–127. doi:10.1101/SQB.1966.031.01.019. PMID4966069.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.