This article has multiple issues. Please help improve it or discuss these issues on the talk page . (Learn how and when to remove these messages)
|

Chiral Lewis acids (CLAs) are a type of Lewis acid catalyst. These acids affect the chirality of the substrate as they react with it. In such reactions, synthesis favors the formation of a specific enantiomer or diastereomer. The method is an enantioselective asymmetric synthesis reaction. Since they affect chirality, they produce optically active products from optically inactive or mixed starting materials. This type of preferential formation of one enantiomer or diastereomer over the other is formally known as asymmetric induction. In this kind of Lewis acid, the electron-accepting atom is typically a metal, such as indium, zinc, lithium, aluminium, titanium, or boron. The chiral-altering ligands employed for synthesizing these acids often have multiple Lewis basic sites (often a diol or a dinitrogen structure) that allow the formation of a ring structure involving the metal atom. [1] [2]
Contents
- Theory
- Asymmetric synthesis
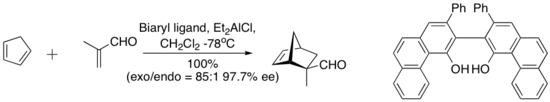
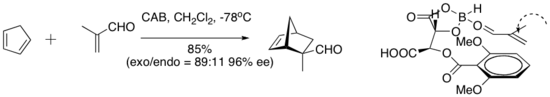
- Diels–Alder reaction
- Aldol reaction
- Baylis–Hillman reaction
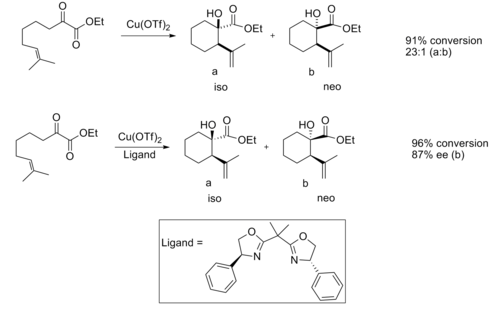
- Ene reaction
- Achiral Lewis acids in stereoselective synthesis
- References
Achiral Lewis acids have been used for decades to promote the synthesis of racemic mixtures in myriad different reactions. Since the 1960s, chemists have used Chiral Lewis acids to induce enantioselective reactions. This is useful when the desired product is a specific enantiomer, as is common in drug synthesis. Common reaction types include Diels–Alder reactions, the ene reaction, [2+2] cycloaddition reactions, hydrocyanation of aldehydes, and most notably, Sharpless epoxidations. [3]