Mutagenesis is a process by which the genetic information of an organism is changed by the production of a mutation. It may occur spontaneously in nature, or as a result of exposure to mutagens. It can also be achieved experimentally using laboratory procedures. A mutagen is a mutation-causing agent, be it chemical or physical, which results in an increased rate of mutations in an organism's genetic code. In nature mutagenesis can lead to cancer and various heritable diseases, and it is also a driving force of evolution. Mutagenesis as a science was developed based on work done by Hermann Muller, Charlotte Auerbach and J. M. Robson in the first half of the 20th century.

In chemistry and biology, reactive oxygen species (ROS) are highly reactive chemicals formed from diatomic oxygen (O2), water, and hydrogen peroxide. Some prominent ROS are hydroperoxide (O2H), superoxide (O2-), hydroxyl radical (OH.), and singlet oxygen. ROS are pervasive because they are readily produced from O2, which is abundant. ROS are important in many ways, both beneficial and otherwise. ROS function as signals, that turn on and off biological functions. They are intermediates in the redox behavior of O2, which is central to fuel cells. ROS are central to the photodegradation of organic pollutants in the atmosphere. Most often however, ROS are discussed in a biological context, ranging from their effects on aging and their role in causing dangerous genetic mutations.

DNA repair is a collection of processes by which a cell identifies and corrects damage to the DNA molecules that encode its genome. In human cells, both normal metabolic activities and environmental factors such as radiation can cause DNA damage, resulting in tens of thousands of individual molecular lesions per cell per day. Many of these lesions cause structural damage to the DNA molecule and can alter or eliminate the cell's ability to transcribe the gene that the affected DNA encodes. Other lesions induce potentially harmful mutations in the cell's genome, which affect the survival of its daughter cells after it undergoes mitosis. As a consequence, the DNA repair process is constantly active as it responds to damage in the DNA structure. When normal repair processes fail, and when cellular apoptosis does not occur, irreparable DNA damage may occur. This can eventually lead to malignant tumors, or cancer as per the two-hit hypothesis.
Lipid peroxidation, or lipid oxidation, is a complex chemical process that leads to oxidative degradation of lipids, resulting in the formation of peroxide and hydroperoxide derivatives. It occurs when free radicals, specifically reactive oxygen species (ROS), interact with lipids within cell membranes, typically polyunsaturated fatty acids (PUFAs) as they have carbon–carbon double bonds. This reaction leads to the formation of lipid radicals, collectively referred to as lipid peroxides or lipid oxidation products (LOPs), which in turn react with other oxidizing agents, leading to a chain reaction that results in oxidative stress and cell damage.

Cockayne syndrome (CS), also called Neill-Dingwall syndrome, is a rare and fatal autosomal recessive neurodegenerative disorder characterized by growth failure, impaired development of the nervous system, abnormal sensitivity to sunlight (photosensitivity), eye disorders and premature aging. Failure to thrive and neurological disorders are criteria for diagnosis, while photosensitivity, hearing loss, eye abnormalities, and cavities are other very common features. Problems with any or all of the internal organs are possible. It is associated with a group of disorders called leukodystrophies, which are conditions characterized by degradation of neurological white matter. There are two primary types of Cockayne syndrome: Cockayne syndrome type A (CSA), arising from mutations in the ERCC8 gene, and Cockayne syndrome type B (CSB), resulting from mutations in the ERCC6 gene.

Nucleotide excision repair is a DNA repair mechanism. DNA damage occurs constantly because of chemicals, radiation and other mutagens. Three excision repair pathways exist to repair single stranded DNA damage: Nucleotide excision repair (NER), base excision repair (BER), and DNA mismatch repair (MMR). While the BER pathway can recognize specific non-bulky lesions in DNA, it can correct only damaged bases that are removed by specific glycosylases. Similarly, the MMR pathway only targets mismatched Watson-Crick base pairs.

Oxidative stress reflects an imbalance between the systemic manifestation of reactive oxygen species and a biological system's ability to readily detoxify the reactive intermediates or to repair the resulting damage. Disturbances in the normal redox state of cells can cause toxic effects through the production of peroxides and free radicals that damage all components of the cell, including proteins, lipids, and DNA. Oxidative stress from oxidative metabolism causes base damage, as well as strand breaks in DNA. Base damage is mostly indirect and caused by the reactive oxygen species generated, e.g., O−
2, OH and H2O2. Further, some reactive oxidative species act as cellular messengers in redox signaling. Thus, oxidative stress can cause disruptions in normal mechanisms of cellular signaling.
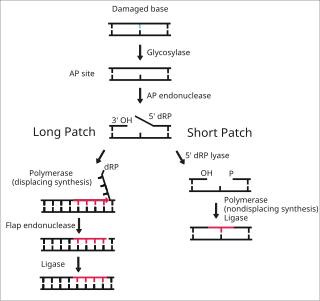
Base excision repair (BER) is a cellular mechanism, studied in the fields of biochemistry and genetics, that repairs damaged DNA throughout the cell cycle. It is responsible primarily for removing small, non-helix-distorting base lesions from the genome. The related nucleotide excision repair pathway repairs bulky helix-distorting lesions. BER is important for removing damaged bases that could otherwise cause mutations by mispairing or lead to breaks in DNA during replication. BER is initiated by DNA glycosylases, which recognize and remove specific damaged or inappropriate bases, forming AP sites. These are then cleaved by an AP endonuclease. The resulting single-strand break can then be processed by either short-patch or long-patch BER.
DNA oxidation is the process of oxidative damage of deoxyribonucleic acid. As described in detail by Burrows et al., 8-oxo-2'-deoxyguanosine (8-oxo-dG) is the most common oxidative lesion observed in duplex DNA because guanine has a lower one-electron reduction potential than the other nucleosides in DNA. The one electron reduction potentials of the nucleosides are guanine 1.29, adenine 1.42, cytosine 1.6 and thymine 1.7. About 1 in 40,000 guanines in the genome are present as 8-oxo-dG under normal conditions. This means that >30,000 8-oxo-dGs may exist at any given time in the genome of a human cell. Another product of DNA oxidation is 8-oxo-dA. 8-oxo-dA occurs at about 1/10 the frequency of 8-oxo-dG. The reduction potential of guanine may be reduced by as much as 50%, depending on the particular neighboring nucleosides stacked next to it within DNA.
Cell damage is a variety of changes of stress that a cell suffers due to external as well as internal environmental changes. Amongst other causes, this can be due to physical, chemical, infectious, biological, nutritional or immunological factors. Cell damage can be reversible or irreversible. Depending on the extent of injury, the cellular response may be adaptive and where possible, homeostasis is restored. Cell death occurs when the severity of the injury exceeds the cell's ability to repair itself. Cell death is relative to both the length of exposure to a harmful stimulus and the severity of the damage caused. Cell death may occur by necrosis or apoptosis.
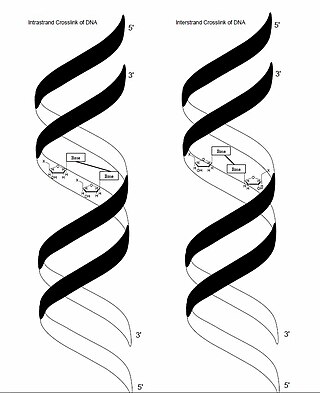
In genetics, crosslinking of DNA occurs when various exogenous or endogenous agents react with two nucleotides of DNA, forming a covalent linkage between them. This crosslink can occur within the same strand (intrastrand) or between opposite strands of double-stranded DNA (interstrand). These adducts interfere with cellular metabolism, such as DNA replication and transcription, triggering cell death. These crosslinks can, however, be repaired through excision or recombination pathways.

In molecular genetics, a DNA adduct is a segment of DNA bound to a cancer-causing chemical. This process could lead to the development of cancerous cells, or carcinogenesis. DNA adducts in scientific experiments are used as biomarkers of exposure. They are especially useful in quantifying an organism's exposure to a carcinogen. The presence of such an adduct indicates prior exposure to a potential carcinogen, but it does not necessarily indicate the presence of cancer in the subject animal.

TFIIH subunit XPD is a protein that in humans is encoded by the ERCC2 gene. It is a component of the general transcription and DNA repair factor IIH (TFIIH) core complex involved in transcription-coupled nucleotide excision repair.

Xeroderma pigmentosum, complementation group C, also known as XPC, is a protein which in humans is encoded by the XPC gene. XPC is involved in the recognition of bulky DNA adducts in nucleotide excision repair. It is located on chromosome 3.
At its simplest, the adductome is the totality of chemical adducts that are present in particular cellular macromolecules such as DNA, and RNA, or proteins found within the organism. These adducts can detrimentally alter the chemical properties of these macromolecules and are therefore also referred to as damage. Adducts may arise as a consequence of the chemical reaction between a given "physicochemical agent to which an organism is exposed across the lifespan". These physicochemical agents can be exogenous in origin, and include ionizing and non-ionizing radiation, the diet, lifestyle factors, pollution, and xenobiotics. They made damage the macromolecules directly, or indirectly e.g., some xenobiotic substances require metabolism of the xenobiotic to produce a chemically reactive metabolite which can then form a covalent bond with the endogenous macromolecule. Agents that damage macromolecules can also arise from endogenous sources, such as reactive oxygen species that are a side product of normal respiration, leading to the formation of oxidatively damaged DNA etc., or other reactive species e.g., reactive nitrogen, sulphur, carbon, selenium and halogen species.
The DNA damage theory of aging proposes that aging is a consequence of unrepaired accumulation of naturally occurring DNA damage. Damage in this context is a DNA alteration that has an abnormal structure. Although both mitochondrial and nuclear DNA damage can contribute to aging, nuclear DNA is the main subject of this analysis. Nuclear DNA damage can contribute to aging either indirectly or directly.

8-Oxo-2'-deoxyguanosine (8-oxo-dG) is an oxidized derivative of deoxyguanosine. 8-Oxo-dG is one of the major products of DNA oxidation. Concentrations of 8-oxo-dG within a cell are a measurement of oxidative stress.
Arsenic biochemistry refers to biochemical processes that can use arsenic or its compounds, such as arsenate. Arsenic is a moderately abundant element in Earth's crust, and although many arsenic compounds are often considered highly toxic to most life, a wide variety of organoarsenic compounds are produced biologically and various organic and inorganic arsenic compounds are metabolized by numerous organisms. This pattern is general for other related elements, including selenium, which can exhibit both beneficial and deleterious effects. Arsenic biochemistry has become topical since many toxic arsenic compounds are found in some aquifers, potentially affecting many millions of people via biochemical processes.
DNA damage is an alteration in the chemical structure of DNA, such as a break in a strand of DNA, a nucleobase missing from the backbone of DNA, or a chemically changed base such as 8-OHdG. DNA damage can occur naturally or via environmental factors, but is distinctly different from mutation, although both are types of error in DNA. DNA damage is an abnormal chemical structure in DNA, while a mutation is a change in the sequence of base pairs. DNA damages cause changes in the structure of the genetic material and prevents the replication mechanism from functioning and performing properly. The DNA damage response (DDR) is a complex signal transduction pathway which recognizes when DNA is damaged and initiates the cellular response to the damage.

Cynthia J. Burrows is an American chemist, currently a distinguished professor in the department of chemistry at the University of Utah, where she is also the Thatcher Presidential Endowed Chair of Biological Chemistry. Burrows was the Senior Editor of the Journal of Organic Chemistry (2001-2013) and became Editor-in-Chief of Accounts of Chemical Research in 2014.,,




