
Succinate dehydrogenase complex subunit C, also known as succinate dehydrogenase cytochrome b560 subunit, mitochondrial, is a protein that in humans is encoded by the SDHC gene. This gene encodes one of four nuclear-encoded subunits that comprise succinate dehydrogenase, also known as mitochondrial complex II, a key enzyme complex of the tricarboxylic acid cycle and aerobic respiratory chains of mitochondria. The encoded protein is one of two integral membrane proteins that anchor other subunits of the complex, which form the catalytic core, to the inner mitochondrial membrane. There are several related pseudogenes for this gene on different chromosomes. Mutations in this gene have been associated with pheochromocytomas and paragangliomas. Alternatively spliced transcript variants have been described.

Succinate dehydrogenase [ubiquinone] iron-sulfur subunit, mitochondrial (SDHB) also known as iron-sulfur subunit of complex II (Ip) is a protein that in humans is encoded by the SDHB gene.

Succinate dehydrogenase complex, subunit A, flavoprotein variant is a protein that in humans is encoded by the SDHA gene. This gene encodes a major catalytic subunit of succinate-ubiquinone oxidoreductase, a complex of the mitochondrial respiratory chain. The complex is composed of four nuclear-encoded subunits and is localized in the mitochondrial inner membrane. SDHA contains the FAD binding site where succinate is deprotonated and converted to fumarate. Mutations in this gene have been associated with a form of mitochondrial respiratory chain deficiency known as Leigh Syndrome. A pseudogene has been identified on chromosome 3q29. Alternatively spliced transcript variants encoding different isoforms have been found for this gene.

Cytochrome b is a protein that in humans is encoded by the MT-CYB gene. Its gene product is a subunit of the respiratory chain protein ubiquinol–cytochrome c reductase, which consists of the products of one mitochondrially encoded gene, MT-CYB, and ten nuclear genes—UQCRC1, UQCRC2, CYC1, UQCRFS1, UQCRB, "11kDa protein", UQCRH, Rieske protein presequence, "cyt c1 associated protein", and Rieske-associated protein.
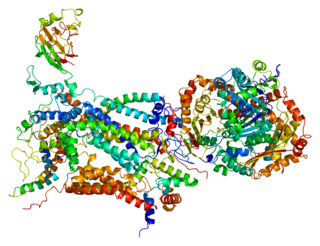
Cytochrome c1, heme protein, mitochondrial (CYC1), also known as UQCR4, MC3DN6, Complex III subunit 4, Cytochrome b-c1 complex subunit 4, or Ubiquinol-cytochrome-c reductase complex cytochrome c1 subunit is a protein that in humans is encoded by the CYC1 gene. CYC1 is a respiratory subunit of Ubiquinol Cytochrome c Reductase, which is located in the inner mitochondrial membrane and is part of the electron transport chain. Mutations in this gene may cause mitochondrial complex III deficiency, nuclear, type 6.

NADH dehydrogenase [ubiquinone] iron-sulfur protein 4, mitochondrial (NDUFS4) also known as NADH-ubiquinone oxidoreductase 18 kDa subunit is an enzyme that in humans is encoded by the NDUFS4 gene. This gene encodes an nuclear-encoded accessory subunit of the mitochondrial membrane respiratory chain NADH dehydrogenase. Complex I removes electrons from NADH and passes them to the electron acceptor ubiquinone. Mutations in this gene can cause mitochondrial complex I deficiencies such as Leigh syndrome.

NADH dehydrogenase [ubiquinone] iron-sulfur protein 3, mitochondrial is an enzyme that in humans is encoded by the NDUFS3 gene on chromosome 11. This gene encodes one of the iron-sulfur protein (IP) components of mitochondrial NADH:ubiquinone oxidoreductase. Mutations in this gene are associated with Leigh syndrome resulting from mitochondrial complex I deficiency.
NADH dehydrogenase [ubiquinone] iron-sulfur protein 8, mitochondrial also known as NADH-ubiquinone oxidoreductase 23 kDa subunit, Complex I-23kD (CI-23kD), or TYKY subunit is an enzyme that in humans is encoded by the NDUFS8 gene. The NDUFS8 protein is a subunit of NADH dehydrogenase (ubiquinone) also known as Complex I, which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain. Mutations in this gene have been associated with Leigh syndrome.

NADH dehydrogenase [ubiquinone] iron-sulfur protein 2, mitochondrial (NDUFS2) also known as NADH-ubiquinone oxidoreductase 49 kDa subunit is an enzyme that in humans is encoded by the NDUFS2 gene. The protein encoded by this gene is a core subunit of the mitochondrial membrane respiratory chain NADH dehydrogenase. Mutations in this gene are associated with mitochondrial complex I deficiency.

NADH-ubiquinone oxidoreductase 75 kDa subunit, mitochondrial (NDUFS1) is an enzyme that in humans is encoded by the NDUFS1 gene. The encoded protein, NDUFS1, is the largest subunit of complex I, located on the inner mitochondrial membrane, and is important for mitochondrial oxidative phosphorylation. Mutations in this gene are associated with complex I deficiency.

NADH dehydrogenase [ubiquinone] flavoprotein 2, mitochondrial (NDUFV2) is an enzyme that in humans is encoded by the NDUFV2 gene. The encoded protein, NDUFV2, is a subunit of complex I of the mitochondrial respiratory chain, which is located on the inner mitochondrial membrane and involved in oxidative phosphorylation. Mutations in this gene are implicated in Parkinson's disease, bipolar disorder, schizophrenia, and have been found in one case of early onset hypertrophic cardiomyopathy and encephalopathy.

NADH dehydrogenase [ubiquinone] iron-sulfur protein 7, mitochondrial, also knowns as NADH-ubiquinone oxidoreductase 20 kDa subunit, Complex I-20kD (CI-20kD), or PSST subunit is an enzyme that in humans is encoded by the NDUFS7 gene. The NDUFS7 protein is a subunit of NADH dehydrogenase (ubiquinone) also known as Complex I, which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain.

NADH dehydrogenase [ubiquinone] 1 beta subcomplex subunit 6, also known as complex I-B17, is a protein that in humans is encoded by the NDUFB6 gene. NADH dehydrogenase (ubiquinone) 1 beta subcomplex subunit 6, is an accessory subunit of the NADH dehydrogenase (ubiquinone) complex, located in the mitochondrial inner membrane. It is also known as Complex I and is the largest of the five complexes of the electron transport chain.
NADH dehydrogenase [ubiquinone] iron-sulfur protein 6, mitochondrial is an enzyme that in humans is encoded by the NDUFS6 gene.
FAD-dependent oxidoreductase domain-containing protein 1 (FOXRED1), also known as H17, or FP634 is an enzyme that in humans is encoded by the FOXRED1 gene. FOXRED1 is an oxidoreductase and complex I-specific molecular chaperone involved in the assembly and stabilization of NADH dehydrogenase (ubiquinone) also known as complex I, which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain. Mutations in FOXRED1 have been associated with Leigh syndrome and infantile-onset mitochondrial encephalopathy.
NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 8 is an enzyme that in humans is encoded by the NDUFA8 gene. The NDUFA8 protein is a subunit of NADH dehydrogenase (ubiquinone), which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain.

Cytochrome c oxidase assembly protein COX15 homolog (COX15), also known as heme A synthase, is a protein that in humans is encoded by the COX15 gene. This protein localizes to the inner mitochondrial membrane and involved in heme A biosynthesis. COX15 is also part of a three-component mono-oxygenase that catalyses the hydroxylation of the methyl group at position eight of the protoheme molecule. Mutations in this gene has been reported in patients with hypertrophic cardiomyopathy as well as Leigh syndrome, and characterized by delayed onset of symptoms, hypotonia, feeding difficulties, failure to thrive, motor regression, and brain stem signs.

NADH:ubiquinone oxidoreductase complex assembly factor 4, (NDUFAF4) also known as Hormone-regulated proliferation-associated protein of 20 kDa, (HRPAP20) or C6orf66 is a protein that in humans is encoded by the NDUFAF4 gene. NDUFAF4 is a mitochondrial assembly protein involved in the assembly of NADH dehydrogenase (ubiquinone) also known as complex I, which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain. Mutations in this gene have been associated with complex I deficiency and infantile mitochondrial encephalomyopathy. Elevations in HRPAP20 have also been implicated in breast cancer.
Protein arginine methyltransferase NDUFAF7, mitochondrial, also known as NADH:ubiquinone oxidoreductase complex assembly factor 7 (NDUFAF7),MidA, C2orf56, or PRO1853, is a protein that in humans is encoded by the NDUFAF7 gene. NDUFAF7 is a methyltransferase mitochondrial assembly enzyme involved in the assembly and stabilization of NADH dehydrogenase (ubiquinone) also known as complex I, which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain. Mutations in NDUFAF7 have been associated with pathologic myopia and complex I deficiency.

Transmembrane protein 70 is a protein that in humans is encoded by the TMEM70 gene. It is a transmembrane protein located in the mitochondrial inner membrane involved in the assembly of the F1 and Fo structural subunits of ATP synthase. Mutations in this gene have been associated with neonatal mitochondrial encephalo-cardiomyopathy due to ATP synthase deficiency, causing a wide variety of symptoms including 3-methylglutaconic aciduria, lactic acidosis, mitochondrial myopathy, and cardiomyopathy.



