
Panniculitis is a group of diseases whose hallmark is inflammation of subcutaneous adipose tissue. Symptoms include tender skin nodules, and systemic signs such as weight loss and fatigue.

Arthrogryposis (AMC) describes congenital joint contracture in two or more areas of the body. It derives its name from Greek, literally meaning 'curving of joints'.

Aase syndrome or Aase–Smith syndrome is a rare inherited disorder characterized by anemia with some joint and skeletal deformities. Aase syndrome is thought to be an autosomal dominant inherited disorder. The genetic basis of the disease is not known. The anemia is caused by underdevelopment of the bone marrow, which is where blood cells are formed.

Behr syndrome is characterized by the association of early-onset optic atrophy with spinocerebellar degeneration resulting in ataxia, pyramidal signs, peripheral neuropathy and developmental delay.
Farber disease is an extremely rare, progressive, autosomal recessive lysosomal storage disease caused by a deficiency of the acid ceramidase enzyme. Acid ceramidase is responsible for breaking down ceramide into sphingosine and fatty acid. When the enzyme is deficient, this leads to an accumulation of fatty material in the lysosomes of the cells, leading to the signs and symptoms of this disorder.
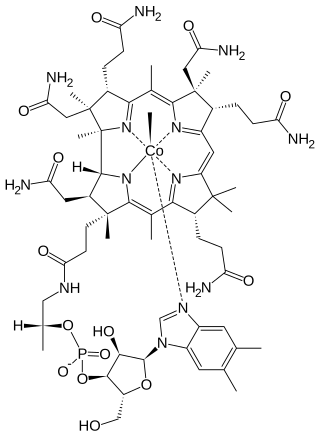
Arakawa's syndrome II is an autosomal dominant metabolic disorder that causes a deficiency of the enzyme tetrahydrofolate-methyltransferase; affected individuals cannot properly metabolize methylcobalamin, a type of Vitamin B12.

Micrognathism is a condition where the jaw is undersized. It is also sometimes called mandibular hypoplasia. It is common in infants, but is usually self-corrected during growth, due to the jaws' increasing in size. It may be a cause of abnormal tooth alignment and in severe cases can hamper feeding. It can also, both in adults and children, make intubation difficult, either during anesthesia or in emergency situations.
An immunoproteasome is a type of proteasome that degrades ubiquitin-labeled proteins found in the cytoplasm in cells exposed to oxidative stress and proinflammatory stimuli. In general, proteasomes consist of a regulatory and a catalytic part. Immunoproteasomes are induced by interferon gamma and oxidative stress, which in the cell triggers the transcription of three catalytic subunits that do not occur in the classical proteasome. Another possible variation of proteasome is the thymoproteasome, which is located in the thymus and folds to present peptides to naive T cells.

Laminopathies are a group of rare genetic disorders caused by mutations in genes encoding proteins of the nuclear lamina. Since the first reports of laminopathies in the late 1990s, increased research efforts have started to uncover the vital role of nuclear envelope proteins in cell and tissue integrity in animals. Laminopathies are a group of degenerative diseases, other disorders associated with inner nuclear membrane proteins are known as nuclear envelopathies.
Bethlem myopathy is predominantly an autosomal dominant myopathy, classified as a congenital form of limb-girdle muscular dystrophy. There are two types of Bethlem myopathy, based on which type of collagen is affected.

Marden–Walker syndrome (MWS) is a rare autosomal recessive congenital disorder. It is characterized by blepharophimosis, microcephaly, micrognathia, multiple joint contractures, arachnodactyly, camptodactyly, kyphoscoliosis and delayed motor development and is often associated with cystic dysplastic kidneys, dextrocardia, Dandy–Walker malformation and agenesis of corpus callosum.

X-linked spinal muscular atrophy type 2, also known as arthrogryposis multiplex congenita X-linked type 1 (AMCX1), is a rare neurological disorder involving death of motor neurons in the anterior horn of spinal cord resulting in generalised muscle wasting (atrophy). The disease is caused by a mutation in UBA1 gene and is passed in an X-linked recessive manner by carrier mothers to affected sons.
Lethal congenital contracture syndrome 1 (LCCS1), also called multiple contracture syndrome, Finnish type, is an autosomal recessive genetic disorder characterized by total immobility of a fetus, detectable at around the 13th week of pregnancy. LCCS1 invariably leads to prenatal death before the 32nd gestational week. LCCS1 is one of 40 Finnish heritage diseases. It was first described in 1985 and since then, approximately 70 cases have been diagnosed.
Haim–Munk syndrome is a skin disease caused, like Papillon–Lefèvre syndrome, by a mutation in the cathepsin C gene. One of its features is thick curved finger and toenails.

Roberts syndrome, or sometimes called pseudothalidomide syndrome, is an extremely rare autosomal recessive genetic disorder that is characterized by mild to severe prenatal retardation or disruption of cell division, leading to malformation of the bones in the skull, face, arms, and legs.
Majeed syndrome is an inherited skin disorder characterized by chronic recurrent multifocal osteomyelitis, congenital dyserythropoietic anemia and a neutrophilic dermatosis. It is classified as an autoinflammatory bone disorder. The condition is found in people with two defective copies of the LPIN2 gene. LPIN2 encodes lipin-2 which is involved in lipid metabolism. The pathogenesis of this mutation with the clinical manifestations has not been elucidated.
Carrier testing is a type of genetic testing that is used to determine if a person is a carrier for specific autosomal recessive diseases. This kind of testing is used most often by couples who are considering becoming pregnant to determine the risks of their child inheriting one of these genetic disorders.
Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome is an autosomal recessive disorder that presents itself via various autoinflammatory responses throughout the body, multiple types of skin lesions, and recurrent long-term fever symptoms. The current known cause for the disorder is a mutation in the PSMB8 gene or mutations in other closely related genes. The syndrome was first named and classified in March 2010 after four patients were reviewed with similar symptoms. There have been approximately 30 cases reported in the scientific literature as of 2015.
