
Rickets, scientific nomenclature: rachitis, is a condition that results in weak or soft bones in children and is caused by either dietary deficiency or genetic causes. Symptoms include bowed legs, stunted growth, bone pain, large forehead, and trouble sleeping. Complications may include bone deformities, bone pseudofractures and fractures, muscle spasms, or an abnormally curved spine. The analogous condition in adults is osteomalacia.

Parathyroid hormone (PTH), also called parathormone or parathyrin, is a peptide hormone secreted by the parathyroid glands that regulates the serum calcium concentration through its effects on bone, kidney, and intestine.
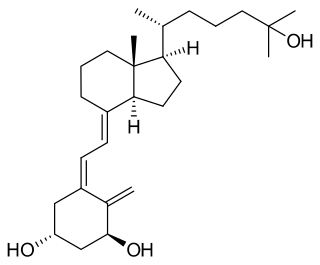
Osteomalacia is a disease characterized by the softening of the bones caused by impaired bone metabolism primarily due to inadequate levels of available phosphate, calcium, and vitamin D, or because of resorption of calcium. The impairment of bone metabolism causes inadequate bone mineralization.

Hyperparathyroidism is an increase in parathyroid hormone (PTH) levels in the blood. This occurs from a disorder either within the parathyroid glands or as response to external stimuli. Symptoms of hyperparathyroidism are caused by inappropriately normal or elevated blood calcium excreted from the bones and flowing into the blood stream in response to increased production of parathyroid hormone. In healthy people, when blood calcium levels are high, parathyroid hormone levels should be low. With long-standing hyperparathyroidism, the most common symptom is kidney stones. Other symptoms may include bone pain, weakness, depression, confusion, and increased urination. Both primary and secondary may result in osteoporosis.

Hypophosphatemia is an electrolyte disorder in which there is a low level of phosphate in the blood. Symptoms may include weakness, trouble breathing, and loss of appetite. Complications may include seizures, coma, rhabdomyolysis, or softening of the bones.
Calcitriol is a hormone and the active form of vitamin D, normally made in the kidney. It is also known as 1,25-dihydroxycholecalciferol. It binds to and activates the vitamin D receptor in the nucleus of the cell, which then increases the expression of many genes. Calcitriol increases blood calcium mainly by increasing the uptake of calcium from the intestines.
Renal osteodystrophy is currently defined as an alteration of bone morphology in patients with chronic kidney disease (CKD). It is one measure of the skeletal component of the systemic disorder of chronic kidney disease-mineral and bone disorder (CKD-MBD). The term "renal osteodystrophy" was coined in 1943, 60 years after an association was identified between bone disease and kidney failure.

Hypophosphatasia (; also called deficiency of alkaline phosphatase, phosphoethanolaminuria, or Rathbun's syndrome; sometimes abbreviated HPP) is a rare, and sometimes fatal, inherited metabolic bone disease. Clinical symptoms are heterogeneous, ranging from the rapidly fatal, perinatal variant, with profound skeletal hypomineralization, respiratory compromise or vitamin B6 dependent seizures to a milder, progressive osteomalacia later in life. Tissue non-specific alkaline phosphatase (TNSALP) deficiency in osteoblasts and chondrocytes impairs bone mineralization, leading to rickets or osteomalacia. The pathognomonic finding is subnormal serum activity of the TNSALP enzyme, which is caused by one of 388 genetic mutations identified to date, in the gene encoding TNSALP. Genetic inheritance is autosomal recessive for the perinatal and infantile forms but either autosomal recessive or autosomal dominant in the milder forms.
Magnesium deficiency is an electrolyte disturbance in which there is a low level of magnesium in the body. Symptoms include tremor, poor coordination, muscle spasms, loss of appetite, personality changes, and nystagmus. Complications may include seizures or cardiac arrest such as from torsade de pointes. Those with low magnesium often have low potassium.
Tertiary hyperparathyroidism is a condition involving the overproduction of the hormone, parathyroid hormone, produced by the parathyroid glands. The parathyroid glands are involved in monitoring and regulating blood calcium levels and respond by either producing or ceasing to produce parathyroid hormone.

Fibroblast growth factor 23 (FGF-23) is a protein and member of the fibroblast growth factor (FGF) family which participates in the regulation of phosphate in plasma and vitamin D metabolism. In humans it is encoded by the FGF23 gene. FGF-23 decreases reabsorption of phosphate in the kidney. Mutations in FGF23 can lead to its increased activity, resulting in autosomal dominant hypophosphatemic rickets.

X-linked hypophosphatemia (XLH) is an X-linked dominant form of rickets that differs from most cases of dietary deficiency rickets in that vitamin D supplementation does not cure it. It can cause bone deformity including short stature and genu varum (bow-leggedness). It is associated with a mutation in the PHEX gene sequence (Xp.22) and subsequent inactivity of the PHEX protein. PHEX mutations lead to an elevated circulating (systemic) level of the hormone FGF23 which results in renal phosphate wasting, and local elevations of the mineralization/calcification-inhibiting protein osteopontin in the extracellular matrix of bones and teeth. An inactivating mutation in the PHEX gene results in an increase in systemic circulating FGF23, and a decrease in the enzymatic activity of the PHEX enzyme which normally removes (degrades) mineralization-inhibiting osteopontin protein; in XLH, the decreased PHEX enzyme activity leads to an accumulation of inhibitory osteopontin locally in bones and teeth to block mineralization which, along with renal phosphate wasting, both cause osteomalacia and odontomalacia.

Nephrocalcinosis, once known as Albright's calcinosis after Fuller Albright, is a term originally used to describe the deposition of poorly soluble calcium salts in the renal parenchyma due to hyperparathyroidism. The term nephrocalcinosis is used to describe the deposition of both calcium oxalate and calcium phosphate. It may cause acute kidney injury. It is now more commonly used to describe diffuse, fine, renal parenchymal calcification in radiology. It is caused by multiple different conditions and is determined by progressive kidney dysfunction. These outlines eventually come together to form a dense mass. During its early stages, nephrocalcinosis is visible on x-ray, and appears as a fine granular mottling over the renal outlines. It is most commonly seen as an incidental finding with medullary sponge kidney on an abdominal x-ray. It may be severe enough to cause renal tubular acidosis or even end stage kidney disease, due to disruption of the kidney tissue by the deposited calcium salts.
Oncogenic osteomalacia, also known as tumor-induced osteomalacia or oncogenic hypophosphatemic osteomalacia, is an uncommon disorder resulting in increased renal phosphate excretion, hypophosphatemia and osteomalacia. It may be caused by a phosphaturic mesenchymal tumor. Symptoms typically include crushing fatigue, severe muscle weakness and brain fog due to the low circulating levels of serum phosphate.

Vitamin D deficiency or hypovitaminosis D is a vitamin D level that is below normal. It most commonly occurs in people when they have inadequate exposure to sunlight, particularly sunlight with adequate ultraviolet B rays (UVB). Vitamin D deficiency can also be caused by inadequate nutritional intake of vitamin D; disorders that limit vitamin D absorption; and disorders that impair the conversion of vitamin D to active metabolites, including certain liver, kidney, and hereditary disorders. Deficiency impairs bone mineralization, leading to bone-softening diseases, such as rickets in children. It can also worsen osteomalacia and osteoporosis in adults, increasing the risk of bone fractures. Muscle weakness is also a common symptom of vitamin D deficiency, further increasing the risk of fall and bone fractures in adults. Vitamin D deficiency is associated with the development of schizophrenia.
An endocrine bone disease is a bone disease associated with a disorder of the endocrine system. An example is osteitis fibrosa cystica.
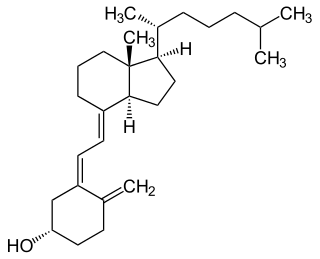
Vitamin D is a group of fat-soluble secosteroids responsible for increasing intestinal absorption of calcium, magnesium, and phosphate, along with numerous other biological functions. In humans, the most significant compounds within this group are vitamin D3 (cholecalciferol) and vitamin D2 (ergocalciferol).
Chronic kidney disease–mineral and bone disorder (CKD–MBD) is one of the many complications associated with chronic kidney disease. It represents a systemic disorder of mineral and bone metabolism due to CKD manifested by either one or a combination of the following:
Burosumab, sold under the brand name Crysvita, is a human monoclonal antibody medication approved 2018 for the treatment of X-linked hypophosphatemia and tumor-induced osteomalacia.

Idiopathic hypercalcinuria (IH) is a condition including an excessive urinary calcium level with a normal blood calcium level resulting from no underlying cause. IH has become the most common cause of hypercalciuria and is the most serious metabolic risk factor for developing nephrolithiasis. IH can predispose individuals to osteopenia or osteoporosis, and affects the entire body. IH arises due to faulty calcium homeostasis, a closely monitored process, where slight deviations in calcium transport in the intestines, blood, and bone can lead to excessive calcium excretion, bone mineral density loss, or kidney stone formation. 50%-60% of nephrolithiasis patients suffer from IH and have 5%-15% lower bone density than those who do not.




