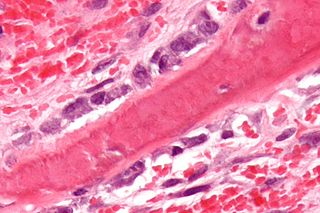
Osteoblasts are cells with a single nucleus that synthesize bone. However, in the process of bone formation, osteoblasts function in groups of connected cells. Individual cells cannot make bone. A group of organized osteoblasts together with the bone made by a unit of cells is usually called the osteon.
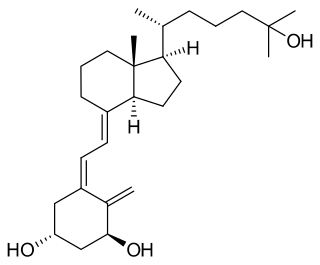
Osteomalacia is a disease characterized by the softening of the bones caused by impaired bone metabolism primarily due to inadequate levels of available phosphate, calcium, and vitamin D, or because of resorption of calcium. The impairment of bone metabolism causes inadequate bone mineralization. Osteomalacia in children is known as rickets, and because of this, use of the term "osteomalacia" is often restricted to the milder, adult form of the disease. Signs and symptoms can include diffuse body pains, muscle weakness, and fragility of the bones. In addition to low systemic levels of circulating mineral ions that result in decreased bone and tooth mineralization, accumulation of mineralization-inhibiting proteins and peptides, and small inhibitory molecules, can occur in the extracellular matrix of bones and teeth, contributing locally to cause matrix hypomineralization (osteomalacia/odontomalacia). A relationship describing local, physiologic double-negative regulation of mineralization has been termed the Stenciling Principle of mineralization, whereby enzyme-substrate pairs imprint mineralization patterns into the extracellular matrix by degrading mineralization inhibitors. The Stenciling Principle for mineralization is particularly relevant to the osteomalacia and odontomalacia observed in hypophosphatasia (HPP) and X-linked hypophosphatemia (XLH).

Hypophosphatemia is an electrolyte disorder in which there is a low level of phosphate in the blood. Symptoms may include weakness, trouble breathing, and loss of appetite. Complications may include seizures, coma, rhabdomyolysis, or softening of the bones.

An osteocyte, an oblate shaped type of bone cell with dendritic processes, is the most commonly found cell in mature bone tissue, and can live as long as the organism itself. The adult human body has about 42 billion of them. Osteocytes do not divide and have an average half life of 25 years. They are derived from osteoprogenitor cells, some of which differentiate into active osteoblasts. Osteoblasts/osteocytes develop in mesenchyme.

Hypophosphatasia (; also called deficiency of alkaline phosphatase, phosphoethanolaminuria, or Rathbun's syndrome; sometimes abbreviated HPP) is a rare, and sometimes fatal, inherited metabolic bone disease. Clinical symptoms are heterogeneous, ranging from the rapidly fatal, perinatal variant, with profound skeletal hypomineralization, respiratory compromise or vitamin B6 dependent seizures to a milder, progressive osteomalacia later in life. Tissue non-specific alkaline phosphatase (TNSALP) deficiency in osteoblasts and chondrocytes impairs bone mineralization, leading to rickets or osteomalacia. The pathognomonic finding is subnormal serum activity of the TNSALP enzyme, which is caused by one of 388 genetic mutations identified to date, in the gene encoding TNSALP. Genetic inheritance is autosomal recessive for the perinatal and infantile forms but either autosomal recessive or autosomal dominant in the milder forms.

Osteopontin (OPN), also known as bone /sialoprotein I, early T-lymphocyte activation (ETA-1), secreted phosphoprotein 1 (SPP1), 2ar and Rickettsia resistance (Ric), is a protein that in humans is encoded by the SPP1 gene. The murine ortholog is Spp1. Osteopontin is a SIBLING (glycoprotein) that was first identified in 1986 in osteoblasts.

Fibroblast growth factor 23 (FGF23) is a protein that in humans is encoded by the FGF23 gene. FGF23 is a member of the fibroblast growth factor (FGF) family which participates in phosphate and vitamin D metabolism and regulation.

X-linked hypophosphatemia (XLH) is an X-linked dominant form of rickets that differs from most cases of dietary deficiency rickets in that vitamin D supplementation does not cure it. It can cause bone deformity including short stature and genu varum (bow-leggedness). It is associated with a mutation in the PHEX gene sequence (Xp.22) and subsequent inactivity of the PHEX protein. PHEX mutations lead to an elevated circulating (systemic) level of the hormone FGF23 which results in renal phosphate wasting, and locally in the extracellular matrix of bones and teeth an elevated level of the mineralization/calcification-inhibiting protein osteopontin. An inactivating mutation in the PHEX gene results in an increase in systemic circulating FGF23, and a decrease in the enzymatic activity of the PHEX enzyme which normally removes (degrades) mineralization-inhibiting osteopontin protein; in XLH, the decreased PHEX enzyme activity leads to an accumulation of inhibitory osteopontin locally in bones and teeth to block mineralization which, along with renal phosphate wasting, both cause osteomalacia and odontomalacia. For both XLH and hypophosphatasia, inhibitor-enzyme pair relationships function to regulate mineralization in the extracellular matrix through a double-negative activation effect in a manner described as the Stenciling Principle. Both these underlying mechanisms contribute to the pathophysiology of XLH that leads to soft bones and teeth. The prevalence of the disease is 1 in 20,000.

Osteonectin (ON) also known as secreted protein acidic and rich in cysteine (SPARC) or basement-membrane protein 40 (BM-40) is a protein that in humans is encoded by the SPARC gene.

Bone sialoprotein (BSP) is a component of mineralized tissues such as bone, dentin, cementum and calcified cartilage. BSP is a significant component of the bone extracellular matrix and has been suggested to constitute approximately 8% of all non-collagenous proteins found in bone and cementum. BSP, a SIBLING protein, was originally isolated from bovine cortical bone as a 23-kDa glycopeptide with high sialic acid content.
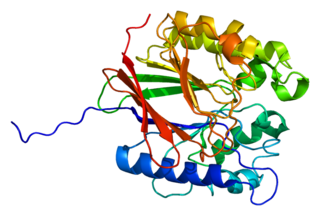
Tartrate-resistant acid phosphatase, also called acid phosphatase 5, tartrate resistant (ACP5), is a glycosylated monomeric metalloprotein enzyme expressed in mammals. It has a molecular weight of approximately 35kDa, a basic isoelectric point (7.6–9.5), and optimal activity in acidic conditions. TRAP is synthesized as latent proenzyme and activated by proteolytic cleavage and reduction. It is differentiated from other mammalian acid phosphatases by its resistance to inhibition by tartrate and by its molecular weight.

Growth differentiation factor 6 (GDF6) is a protein that in humans is encoded by the GDF6 gene.

Low-density lipoprotein receptor-related protein 5 is a protein that in humans is encoded by the LRP5 gene. LRP5 is a key component of the LRP5/LRP6/Frizzled co-receptor group that is involved in canonical Wnt pathway. Mutations in LRP5 can lead to considerable changes in bone mass. A loss-of-function mutation causes osteoporosis-pseudoglioma, while a gain-of-function mutation causes drastic increases in bone mass.

alpha-2-HS-glycoprotein also known as fetuin-A is a protein that in humans is encoded by the AHSG gene. Fetuin-A belongs to the fetuin class of plasma binding proteins and is more abundant in fetal than adult blood.

Dentin matrix acidic phosphoprotein 1 is a protein that in humans is encoded by the DMP1 gene.
Matrix extracellular phosphoglycoprotein is a protein that in humans is encoded by the MEPE gene. A conserved RGD motif is found in this protein, and this is potentially involved in integrin recognition.
Oncogenic osteomalacia, also known as oncogenic hypophosphatemic osteomalacia, is an uncommon disorder resulting in increased renal phosphate excretion, hypophosphatemia and osteomalacia. It may be caused by a phosphaturic mesenchymal tumor.

Family with sequence similarity 20, member C also known as FAM20C or DMP4 is a protein which in humans is encoded by the FAM20C gene. Fam20C, a Golgi localized protein kinase, is a serine kinase that phosphorylates both casein and other highly acidic proteins and members of the small integrin-binding ligand, the N-linked glycoproteins (SIBLING) family at the target motif SerXGlu.
The family of non-collagenous proteins known as SIBLING proteins, standing for small integrin-binding ligand, N-linked glycoprotein, are components of the extracellular matrix of bone and dentin. Evidence shows that these proteins play key roles in the mineralization of these tissues.
Carboxypeptidase A6 (CPA6) is a metallocarboxypeptidase enzyme that in humans is encoded by the CPA6 gene. It is highly expressed in the adult mouse olfactory bulb and is broadly expressed in the embryonic brain and other tissues.



