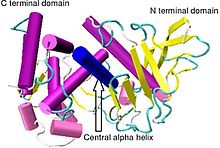
Proteolysis is the breakdown of proteins into smaller polypeptides or amino acids. Uncatalysed, the hydrolysis of peptide bonds is extremely slow, taking hundreds of years. Proteolysis is typically catalysed by cellular enzymes called proteases, but may also occur by intra-molecular digestion.

In biology and biochemistry, the active site is the region of an enzyme where substrate molecules bind and undergo a chemical reaction. The active site consists of amino acid residues that form temporary bonds with the substrate, the binding site, and residues that catalyse a reaction of that substrate, the catalytic site. Although the active site occupies only ~10–20% of the volume of an enzyme, it is the most important part as it directly catalyzes the chemical reaction. It usually consists of three to four amino acids, while other amino acids within the protein are required to maintain the tertiary structure of the enzymes.
Matrix metalloproteinases (MMPs), also known as matrix metallopeptidases or matrixins, are metalloproteinases that are calcium-dependent zinc-containing endopeptidases; other family members are adamalysins, serralysins, and astacins. The MMPs belong to a larger family of proteases known as the metzincin superfamily.
In biology and biochemistry, protease inhibitors, or antiproteases, are molecules that inhibit the function of proteases. Many naturally occurring protease inhibitors are proteins.

A metalloproteinase, or metalloprotease, is any protease enzyme whose catalytic mechanism involves a metal. An example is ADAM12 which plays a significant role in the fusion of muscle cells during embryo development, in a process known as myogenesis.

Papain, also known as papaya proteinase I, is a cysteine protease enzyme present in papaya and mountain papaya. It is the namesake member of the papain-like protease family.
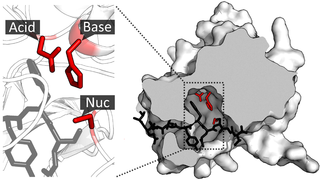
A catalytic triad is a set of three coordinated amino acids that can be found in the active site of some enzymes. Catalytic triads are most commonly found in hydrolase and transferase enzymes. An acid-base-nucleophile triad is a common motif for generating a nucleophilic residue for covalent catalysis. The residues form a charge-relay network to polarise and activate the nucleophile, which attacks the substrate, forming a covalent intermediate which is then hydrolysed to release the product and regenerate free enzyme. The nucleophile is most commonly a serine or cysteine amino acid, but occasionally threonine or even selenocysteine. The 3D structure of the enzyme brings together the triad residues in a precise orientation, even though they may be far apart in the sequence.
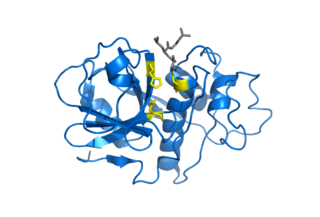
Cysteine proteases, also known as thiol proteases, are hydrolase enzymes that degrade proteins. These proteases share a common catalytic mechanism that involves a nucleophilic cysteine thiol in a catalytic triad or dyad.
Gelatinases are enzymes capable of degrading gelatin through hydrolysis, playing a major role in degradation of extracellular matrix and tissue remodeling. Gelatinases are a type of matrix metalloproteinase (MMP), a family of enzymes that depend on zinc as a cofactor and can break down parts of the extracellular matrix. MMPs have multiple subgroups, including gelatinase A and gelatinase B. Gelatinases are assigned a variety of Enzyme Commission numbers: gelatinase A uses 3.4.24.24, and gelatinase B uses 3.4.24.35, in which the first three numbers are same. The first digit, 3, is the class. Class 3 enzymes are hydrolases, enzymes that catalyze hydrolysis reactions, that is, they cleave bonds in presence of water. The next digit represents sub-class 4, or proteases, which are enzymes who hydrolyze peptide bonds in proteins. The next number is the sub-subclass of 24, which consists of metalloendopeptidases which contain metal ions in their active sites, in this case zinc, which help in cleaving peptide bonds. The last part of the EC number is the serial number, identifying specific enzymes within a sub-subclass. 24 represents gelatinase A, which is a metalloproteinase that breaks down gelatin and collagen, while 35 represents gelatinase B, which hydrolyzes peptide bonds.
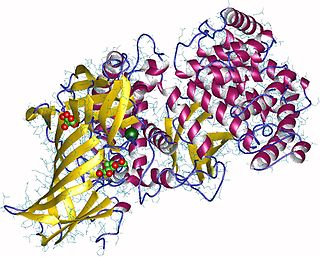
Aminopeptidases are enzymes that catalyze the cleavage of amino acids from the N-terminus (beginning), of proteins or peptides. They are found in many organisms; in the cell, they are found in many organelles, in the cytosol, and as membrane proteins. Aminopeptidases are used in essential cellular functions, and are often zinc metalloenzymes, containing a zinc cofactor.
Transition state analogs, are chemical compounds with a chemical structure that resembles the transition state of a substrate molecule in an enzyme-catalyzed chemical reaction. Enzymes interact with a substrate by means of strain or distortions, moving the substrate towards the transition state. Transition state analogs can be used as inhibitors in enzyme-catalyzed reactions by blocking the active site of the enzyme. Theory suggests that enzyme inhibitors which resembled the transition state structure would bind more tightly to the enzyme than the actual substrate. Examples of drugs that are transition state analog inhibitors include flu medications such as the neuraminidase inhibitor oseltamivir and the HIV protease inhibitors saquinavir in the treatment of AIDS.

Insulin-degrading enzyme, also known as IDE, is an enzyme.

In molecular biology, Proteinase K is a broad-spectrum serine protease. The enzyme was discovered in 1974 in extracts of the fungus Parengyodontium album. Proteinase K is able to digest hair (keratin), hence, the name "Proteinase K". The predominant site of cleavage is the peptide bond adjacent to the carboxyl group of aliphatic and aromatic amino acids with blocked alpha amino groups. It is commonly used for its broad specificity. This enzyme belongs to Peptidase family S8 (subtilisin). The molecular weight of Proteinase K is 28,900 daltons.

The EGF-like domain is an evolutionary conserved protein domain, which derives its name from the epidermal growth factor where it was first described. It comprises about 30 to 40 amino-acid residues and has been found in a large number of mostly animal proteins. Most occurrences of the EGF-like domain are found in the extracellular domain of membrane-bound proteins or in proteins known to be secreted. An exception to this is the prostaglandin-endoperoxide synthase. The EGF-like domain includes 6 cysteine residues which in the epidermal growth factor have been shown to form 3 disulfide bonds. The structures of 4-disulfide EGF-domains have been solved from the laminin and integrin proteins. The main structure of EGF-like domains is a two-stranded β-sheet followed by a loop to a short C-terminal, two-stranded β-sheet. These two β-sheets are usually denoted as the major (N-terminal) and minor (C-terminal) sheets. EGF-like domains frequently occur in numerous tandem copies in proteins: these repeats typically fold together to form a single, linear solenoid domain block as a functional unit.

An Oligopeptidase is an enzyme that cleaves peptides but not proteins. This property is due to its structure: the active site of this enzyme is located at the end of a narrow cavity which can only be reached by peptides.
The Kazal domain is an evolutionary conserved protein domain usually indicative of serine protease inhibitors. However, kazal-like domains are also seen in the extracellular part of agrins, which are not known to be protease inhibitors.
Caricain is an enzyme. This enzyme catalyses the following chemical reaction: Hydrolysis of proteins with broad specificity for peptide bonds, similar to those of papain and chymopapain
Bacillolysin is an enzyme. This enzyme catalyses the following chemical reaction

β-propeller phytases (BPPs) are a group of enzymes (i.e. protein superfamily) with a round beta-propeller structure. BPPs are phytases, which means that they are able to remove (hydrolyze) phosphate groups from phytic acid and its phytate salts. Hydrolysis happens stepwise and usually ends in myo-inositol triphosphate product which has three phosphate groups still bound to it. The actual substrate of BPPs is calcium phytate and in order to hydrolyze it, BPPs must have Ca2+ ions bound to themselves. BPPs are the most widely found phytase superfamily in the environment and they are thought to have a major role in phytate-phosphorus cycling in soil and water. As their alternative name alkaline phytase suggests, BPPs work best in basic (or neutral) environment. Their pH optima is 6–9, which is unique among the phytases.
RopB transcriptional regulator, also known as RopB/Rgg transcriptional regulator is a transcriptional regulator protein that regulates expression of the extracellularly secreted cysteine protease streptococcal pyrogenic exotoxin B (speB) [See Also: erythrogenic toxins] which is an important virulence factor of Streptococcus pyogenes and is responsible for the dissemination of a host of infectious diseases including strep throat, impetigo, streptococcal toxic shock syndrome, necrotizing fasciitis, and scarlet fever. Functional studies suggest that the ropB multigene regulon is responsible for not only global regulation of virulence but also a wide range of functions from stress response, metabolic function, and two-component signaling. Structural studies implicate ropB's regulatory action being reliant on a complex interaction involving quorum sensing with the leaderless peptide signal speB-inducing peptide (SIP) acting in conjunction with a pH sensitive histidine switch.