
Joubert syndrome is a rare autosomal recessive genetic disorder that affects the cerebellum, an area of the brain that controls balance and coordination.

Holoprosencephaly (HPE) is a cephalic disorder in which the prosencephalon fails to develop into two hemispheres, typically occurring between the 18th and 28th day of gestation. Normally, the forebrain is formed and the face begins to develop in the fifth and sixth weeks of human pregnancy. The condition also occurs in other species.

ICF syndrome is a very rare autosomal recessive immune disorder.

X-linked recessive inheritance is a mode of inheritance in which a mutation in a gene on the X chromosome causes the phenotype to be always expressed in males and in females who are homozygous for the gene mutation, see zygosity. Females with one copy of the mutated gene are carriers.
Chédiak–Higashi syndrome (CHS) is a rare autosomal recessive disorder that arises from a mutation of a lysosomal trafficking regulator protein, which leads to a decrease in phagocytosis. The decrease in phagocytosis results in recurrent pyogenic infections, albinism, and peripheral neuropathy.
Severe congenital neutropenia (SCN), also often known as Kostmann syndrome or disease, is a group of rare disorders that affect myelopoiesis, causing a congenital form of neutropenia, usually without other physical malformations. SCN manifests in infancy with life-threatening bacterial infections. It causes severe pyogenic infections. It can be caused by autosomal dominant inheritance of the ELANE gene, autosomal recessive inheritance of the HAX1 gene. There is an increased risk of leukemia and myelodysplastic cancers.
Heřmanský–Pudlák syndrome is an extremely rare autosomal recessive disorder which results in oculocutaneous albinism, bleeding problems due to a platelet abnormality, and storage of an abnormal fat-protein compound. It is thought to affect around 1 in 500,000 people worldwide, with a significantly higher occurrence in Puerto Ricans, with a prevalence of 1 in 1800. Many of the clinical research studies on the disease have been conducted in Puerto Rico.
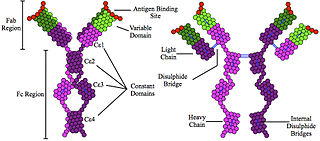
Hyperimmunoglobulinemia E syndrome (HIES), of which the autosomal dominant form is called Job's syndrome or Buckley syndrome, is a heterogeneous group of immune disorders. Job's is also very rare at about 300 cases currently in the literature.

Larsen syndrome (LS) is a congenital disorder discovered in 1950 by Larsen and associates when they observed dislocation of the large joints and face anomalies in six of their patients. Patients with Larsen syndrome normally present with a variety of symptoms, including congenital anterior dislocation of the knees, dislocation of the hips and elbows, flattened facial appearance, prominent foreheads, and depressed nasal bridges. Larsen syndrome can also cause a variety of cardiovascular and orthopedic abnormalities. This rare disorder is caused by a genetic defect in the gene encoding filamin B, a cytoplasmic protein that is important in regulating the structure and activity of the cytoskeleton. The gene that influences the emergence of Larsen syndrome is found in chromosome region, 3p21.1-14.1, a region containing human type VII collagen gene. Larsen syndrome has recently been described as a mesenchyme disorder that affects the connective tissue of an individual. Autosomal dominant and recessive forms of the disorder have been reported, although most cases are autosomal dominant. Reports have found that in Western societies, Larsen syndrome can be found in one in every 100,000 births, but this is most likely an underestimate because the disorder is frequently unrecognized or misdiagnosed.

Ethylmalonic encephalopathy (EE) is a rare autosomal recessive inborn error of metabolism. Patients affected with EE are typically identified shortly after birth, with symptoms including diarrhea, petechiae and seizures. The genetic defect in EE is thought to involve an impairment in the degradation of sulfide intermediates in the body. Hydrogen sulfide then builds up to toxic levels. EE was initially described in 1994. Most cases of EE have been described in individuals of Mediterranean or Arabic origin.

Acrocallosal syndrome is an extremely rare autosomal recessive syndrome characterized by corpus callosum agenesis, polydactyly, multiple dysmorphic features, motor and intellectual disabilities, and other symptoms. The syndrome was first described by Albert Schinzel in 1979. Mutations in KIF7 are causative for ACLS, and mutations in GLI3 are associated with a similar syndrome.
Marden–Walker syndrome (MWS) is a rare autosomal recessive congenital disorder. It is characterized by blepharophimosis, microcephaly, micrognathia, multiple joint contractures, arachnodactyly, camptodactyly, kyphoscoliosis and delayed motor development and is often associated with cystic dysplastic kidneys, dextrocardia, Dandy–Walker malformation and agenesis of corpus callosum.
Griscelli syndrome type 2 is a rare autosomal recessive syndrome characterized by variable cutenous albinism, silver colored metallic looking hair, frequent bacterial or viral infections, neutropenia, and thrombocytopenia.

Neu–Laxova syndrome is a rare autosomal recessive disorder characterized by severe intrauterine growth restriction and multiple congenital malformations. Neu–Laxova syndrome is a very severe disorder, leading to stillbirth or death shortly after birth. It was first described by Dr. Richard Neu in 1971 and Dr. Renata Laxova in 1972 as a lethal disorder in siblings with multiple malformations. Neu–Laxova syndrome is an extremely rare disorder with fewer than 100 cases reported in medical literature.
PGM3 deficiency is a rare genetic disorder of the immune system associated with diminished phosphoglucomutase 3 function. PGM3 is an enzyme which in humans is encoded by gene PGM3. This disorder manifests as severe atopy, immune deficiency, autoimmunity, intellectual disability, and hypomyelination. In 2014, Investigators Atfa Sassi at the Pasteur Institute of Tunis, Sandra Lazaroski at the University Medical Center Freiburg, and Gang Wu at the Imperial College London, identified PGM3 mutations in nine patients from four consanguineous families. In the same year, a researchers from the laboratories of Joshua Milner and Helen Su at the National Institute of Allergy and Infectious Disease at the U.S. National Institutes of Health described PGM3 deficiency in eight additional patients from two families.
MEDNIK syndrome(OMIM#609313), also known as "syndrome de Kamouraska", is a genetic disorder that is caused by mutations to the AP1S1 gene. Transmission of the disease is believed to be autosomal recessive. Symptoms of the syndrome are intellectual disability, enteropathy, deafness, neuropathy, ichthyosis, and keratoderma (MEDNIK). People with MEDNIK syndrome often have a high forehead, upslanting palpebral fissures, a depressed nasal bridge, low-set ears, growth retardation, and brain atrophy apparent upon imaging. The disorder was discovered by Patrick Cossette and his research team from the Université de Montréal. MEDNIK syndrome was initially reported in a few French-Canadian families near Quebec who all shared common ancestors.
Chudley–Mccullough syndrome is a rare genetic disorder which is characterized by bilateral congenital hearing loss associated with brain malformations. It is a type of syndromic deafness.
X-linked complicated corpus callosum dysgenesis is a genetic disorder characterized by dysplasia, hypoplasia or agenesis of the corpus callosum alongside variable intellectual disability and spastic paraplegia. Only 13 cases have been described in medical literature. Transmission is X-linked recessive. It is the mildest subtype of L1 syndrome.
