
The N-methyl-D-aspartatereceptor (also known as the NMDA receptor or NMDAR), is a glutamate receptor and predominantly Ca2+ ion channel found in neurons. The NMDA receptor is one of three types of ionotropic glutamate receptors, the other two being AMPA and kainate receptors. Depending on its subunit composition, its ligands are glutamate and glycine (or D-serine). However, the binding of the ligands is typically not sufficient to open the channel as it may be blocked by Mg2+ ions which are only removed when the neuron is sufficiently depolarized. Thus, the channel acts as a "coincidence detector" and only once both of these conditions are met, the channel opens and it allows positively charged ions (cations) to flow through the cell membrane. The NMDA receptor is thought to be very important for controlling synaptic plasticity and mediating learning and memory functions.

AP5 is a chemical compound used as a biochemical tool to study various cellular processes. It is a selective NMDA receptor antagonist that competitively inhibits the ligand (glutamate) binding site of NMDA receptors. AP5 blocks NMDA receptors in micromolar concentrations.

Dizocilpine (INN), also known as MK-801, is a pore blocker of the NMDA receptor, a glutamate receptor, discovered by a team at Merck in 1982. Glutamate is the brain's primary excitatory neurotransmitter. The channel is normally blocked with a magnesium ion and requires depolarization of the neuron to remove the magnesium and allow the glutamate to open the channel, causing an influx of calcium, which then leads to subsequent depolarization. Dizocilpine binds inside the ion channel of the receptor at several of PCP's binding sites thus preventing the flow of ions, including calcium (Ca2+), through the channel. Dizocilpine blocks NMDA receptors in a use- and voltage-dependent manner, since the channel must open for the drug to bind inside it. The drug acts as a potent anti-convulsant and probably has dissociative anesthetic properties, but it is not used clinically for this purpose because of the discovery of brain lesions, called Olney's lesions (see below), in laboratory rats. Dizocilpine is also associated with a number of negative side effects, including cognitive disruption and psychotic-spectrum reactions. It inhibits the induction of long term potentiation and has been found to impair the acquisition of difficult, but not easy, learning tasks in rats and primates. Because of these effects of dizocilpine, the NMDA receptor pore blocker ketamine is used instead as a dissociative anesthetic in human medical procedures. While ketamine may also trigger temporary psychosis in certain individuals, its short half-life and lower potency make it a much safer clinical option. However, dizocilpine is the most frequently used uncompetitive NMDA receptor antagonist in animal models to mimic psychosis for experimental purposes.

CNQX or cyanquixaline (6-cyano-7-nitroquinoxaline-2,3-dione) is a competitive AMPA/kainate receptor antagonist. Its chemical formula is C9H4N4O4. CNQX is often used in the retina to block the responses of OFF-bipolar cells for electrophysiology recordings.
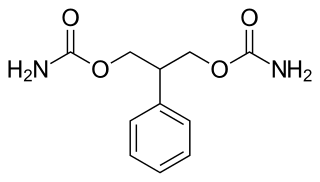
Felbamate is an anticonvulsant used in the treatment of epilepsy. It is used to treat partial seizures in adults and partial and generalized seizures associated with Lennox–Gastaut syndrome in children. However, an increased risk of potentially fatal aplastic anemia and/or liver failure limit the drug's usage to severe refractory epilepsy.

NMDA receptor antagonists are a class of drugs that work to antagonize, or inhibit the action of, the N-Methyl-D-aspartate receptor (NMDAR). They are commonly used as anesthetics for humans and animals; the state of anesthesia they induce is referred to as dissociative anesthesia.

Ifenprodil, sold under the brand names Cerocral, Dilvax, and Vadilex, is a cerebral vasodilator that has been marketed in some countries, including in Japan, Hong Kong, and France. It is currently under development for treatment of a variety of additional indications.

Glutamate [NMDA] receptor subunit epsilon-2, also known as N-methyl D-aspartate receptor subtype 2B, is a protein that in humans is encoded by the GRIN2B gene.

Glutamate [NMDA] receptor subunit 3A is a protein that in humans is encoded by the GRIN3A gene.

Glutamate [NMDA] receptor subunit epsilon-4 is a protein that in humans is encoded by the GRIN2D gene.
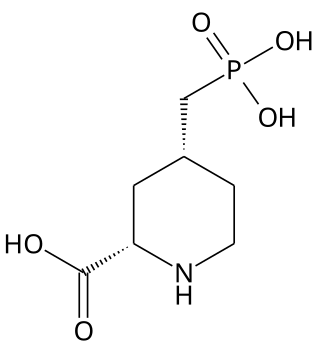
Selfotel (CGS-19755) is a drug which acts as a competitive NMDA antagonist, directly competing with glutamate for binding to the receptor. Initial studies showed it to have anticonvulsant, anxiolytic, analgesic and neuroprotective effects, and it was originally researched for the treatment of stroke, but subsequent animal and human studies showed phencyclidine-like effects, as well as limited efficacy and evidence for possible neurotoxicity under some conditions, and so clinical development was ultimately discontinued.

Neramexane is a drug related to memantine, which acts as an NMDA antagonist and has neuroprotective effects. It is being developed for various possible applications, including treatment of tinnitus, Alzheimer's disease, drug addiction and as an analgesic. Animal studies have also suggested antidepressant and nootropic actions so that this drug may be used for a wide range of potential applications. It also acts as a nicotinic acetylcholine receptor antagonist.
Besonprodil (CI-1041) is a drug which acts as an NMDA antagonist, selective for the NR2B subunit. It is under development as a supplemental medication for Parkinson's disease, and has been shown in animals to be effective in counteracting the dyskinesias associated with long-term treatment with levodopa and related drugs.
Conantokins are a small family of helical peptides that are derived from the venom of predatory marine snails of the genus Conus. Conantokins act as potent and specific antagonists of the N-methyl-D-aspartate receptor (NMDAR). They are the only naturally-derived peptides to do so. The subtypes of conantokins exhibit a surprising variability of selectivity across the NMDAR subunits, and are therefore uniquely useful in developing subunit-specific pharmacological probes.

Rapastinel is a novel antidepressant that was under development by Allergan as an adjunctive therapy for the treatment of treatment-resistant depression. It is a centrally active, intravenously administered amidated tetrapeptide that acts as a novel and selective modulator of the NMDA receptor. The drug is a rapid-acting and long-lasting antidepressant as well as robust cognitive enhancer by virtue of its ability to enhance NMDA receptor-mediated signal transduction and synaptic plasticity.

7-Chlorokynurenic acid (7-CKA) is a tool compound that acts as a potent and selective competitive antagonist of the glycine site of the NMDA receptor. It produces ketamine-like rapid antidepressant effects in animal models of depression. However, 7-CKA is unable to cross the blood-brain-barrier, and for this reason, is unsuitable for clinical use. As a result, a centrally-penetrant prodrug of 7-CKA, 4-chlorokynurenine (AV-101), has been developed for use in humans, and is being studied in clinical trials as a potential treatment for major depressive disorder, and anti-nociception. In addition to antagonizing the NMDA receptor, 7-CKA also acts as a potent inhibitor of the reuptake of glutamate into synaptic vesicles, an action that it mediates via competitive blockade of vesicular glutamate transporters.

Apimostinel is an investigational antidepressant, acting as a novel and selective modulator of the NMDA receptor. It is currently under development for the acute treatment of major depressive disorder (MDD) by Gate Neurosciences, and previously by Naurex and Allergan. As of February 2015, an intravenous formulation of apimostinel has completed a phase IIa clinical trial for MDD.
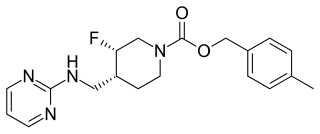
Rislenemdaz is an orally active, selective NMDA receptor subunit 2B (NR2B) antagonist which is under development by Cerecor in the United States as an adjunctive therapy for treatment-resistant depression (TRD). In November 2013, phase II clinical trials were initiated, and in the same month, rislenemdaz received Fast Track Designation from the Food and Drug Administration for TRD.

L-4-Chlorokynurenine is an orally active small molecule prodrug of 7-chlorokynurenic acid, a NMDA receptor antagonist. It was investigated as a potential rapid-acting antidepressant.
