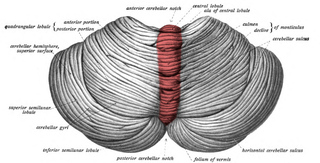
The cerebellar vermis is located in the medial, cortico-nuclear zone of the cerebellum, which is in the posterior fossa of the cranium. The primary fissure in the vermis curves ventrolaterally to the superior surface of the cerebellum, dividing it into anterior and posterior lobes. Functionally, the vermis is associated with bodily posture and locomotion. The vermis is included within the spinocerebellum and receives somatic sensory input from the head and proximal body parts via ascending spinal pathways.

Dandy–Walker malformation (DWM), also known as Dandy–Walker syndrome (DWS), is a rare congenital brain malformation in which the part joining the two hemispheres of the cerebellum does not fully form, and the fourth ventricle and space behind the cerebellum are enlarged with cerebrospinal fluid. Most of those affected develop hydrocephalus within the first year of life, which can present as increasing head size, vomiting, excessive sleepiness, irritability, downward deviation of the eyes and seizures. Other, less common symptoms are generally associated with comorbid genetic conditions and can include congenital heart defects, eye abnormalities, intellectual disability, congenital tumours, other brain defects such as agenesis of the corpus callosum, skeletal abnormalities, an occipital encephalocele or underdeveloped genitalia or kidneys. It is sometimes discovered in adolescents or adults due to mental health problems.
Abruzzo–Erickson syndrome is an extremely rare disorder characterized by deafness, protruding ears, coloboma, a cleft palate or palatal rugosity, radial synostosis, and short stature. It was first characterized by Abruzzo and Erickson in 1977 as a CHARGE like syndrome as variably expressed among a family of two brothers, their mother, and their maternal uncle. Members of this family exhibited many of the CHARGE symptoms, but notably did not have choanal atresia and the brothers experienced typical genital development. Due to the recent discovery of this disorder, its etiology is not fully known but it is understood that it arises from mutations on the TBX22 gene on the X-chromosome. The disorder is inherited in an X-linked recessive manner. There is currently no known cure but its symptoms can be treated.

Micrognathism is a condition where the jaw is undersized. It is also sometimes called mandibular hypoplasia. It is common in infants, but is usually self-corrected during growth, due to the jaws' increasing in size. It may be a cause of abnormal tooth alignment and in severe cases can hamper feeding. It can also, both in adults and children, make intubation difficult, either during anesthesia or in emergency situations.

Young–Simpson syndrome (YSS) is a rare congenital disorder with symptoms including hypothyroidism, heart defects, facial dysmorphism, cryptorchidism in males, hypotonia, intellectual disability, and postnatal growth retardation.

Frontonasal dysplasia (FND) is a congenital malformation of the midface. For the diagnosis of FND, a patient should present at least two of the following characteristics: hypertelorism, a wide nasal root, vertical midline cleft of the nose and/or upper lip, cleft of the wings of the nose, malformed nasal tip, encephalocele or V-shaped hair pattern on the forehead. The cause of FND remains unknown. FND seems to be sporadic (random) and multiple environmental factors are suggested as possible causes for the syndrome. However, in some families multiple cases of FND were reported, which suggests a genetic cause of FND.

Fryns syndrome is an autosomal recessive multiple congenital anomaly syndrome that is usually lethal in the neonatal period. Fryns (1987) reviewed the syndrome.

Roberts syndrome, or sometimes called pseudothalidomide syndrome, is an extremely rare autosomal recessive genetic disorder that is characterized by mild to severe prenatal retardation or disruption of cell division, leading to malformation of the bones in the skull, face, arms, and legs.
Goldberg–Shprintzen is a very rare connective tissue condition associated with mutations in KIAA1279 gene which encodes KIF-binding protein (KBP), a protein that may interact with microtubules and actin filaments. KBP may play a key role in cytoskeleton formation and neurite growth.
13q deletion syndrome is a rare genetic disease caused by the deletion of some or all of the large arm of human chromosome 13. Depending upon the size and location of the deletion on chromosome 13, the physical and mental manifestations will vary. It has the potential to cause intellectual disability and congenital malformations that affect a variety of organ systems. Because of the rarity of the disease in addition to the variations in the disease, the specific genes that cause this disease are unknown. This disease is also known as:
Fryns-Aftimos syndrome is a rare chromosomal condition and is associated with pachygyria, severe mental retardation, epilepsy and characteristic facial features. This syndrome is a malformation syndrome, characterized by numerous facial dysmorphias not limited to hypertelorism, iris or retinal coloboma, cleft lip, and congenital heart defects. This syndrome has been seen in 30 unrelated people. Characterized by a de novo mutation located on chromosome 7p22, there is typically no family history prior to onset. The severity of the disorder can be determined by the size of the deletion on 7p22, enveloping the ACTB gene and surrounding genes, which is consistent with a contiguous gene deletion syndrome. Confirming a diagnosis of Fryns-Aftimos syndrome typically consists of serial single-gene testing or multigene panel of genes of interest or exome sequencing.
Pascual-Castroviejo syndrome type 1 is a rare autosomal recessive condition characterized by facial dysmorphism, cognitive impairment and skeletal anomalies.
Fine–Lubinsky syndrome is a rare genetic disorder which is characterized by ocular and hearing problems, speech and developmental delay, short stature, intellectual disabilities and facial dysmorphisms.
Porencephaly-cerebellar hypoplasia-internal malformations syndrome is a rare autosomal recessive syndrome that mainly affects the central nervous system. It causes cardiac defects, brain anomalies, and craniofacial dysmorphisms. It has been reported in a pair of German siblings of the opposite sex born to consanguineous Turkish parents.

