An artificial metalloenzyme (ArM) is a designer metalloprotein, not found in nature, which can catalyze desired chemical reactions.[1][2] Despite fitting into classical enzyme categories, ArMs also have potential in new-to-nature chemical reactivity like catalysing Suzuki coupling,[3]metathesis[4] etc., which were never reported among natural enzymatic reactions.
ArMs have two main components: a protein scaffold and an artificial catalytic moiety, which, in this case, features a metal center. This class of designer biocatalysts is unique because of the potential to improve the catalytic performance through chemogenetic optimization, a parallel improvement of both the direct metal surrounding (first coordination sphere) and the protein scaffold (second coordination sphere).The second coordination sphere (protein scaffold) is easily evolvable and, in the case of ArMs, responsible for very high (stereo)selectivity.[5] With the progress in organometallic synthesis and protein engineering, more and more new kind of design of ArMs were developed, showing promising future in both academia and industrial aspects.[6]
Dated back to 1956, the first protein modified transition metal catalyst was documented.[7] The Palladium(II) salt was absorbed onto silk fibroin fiber, reduced by hydrogen to get the first reported ArM, which can catalyze asymmetric hydrogenation. This work was not reproducible, but it is considered to be the first work in the field of artificial metalloenzymes.[5] At that time, the major challenge that blocked further studies was underdeveloped protein production and purification technology. The first attempt to anchor an abiotic metal center onto a protein was reported by Whitesideset al. using biotin-avidin interaction, making an artificial hydrogenase.[8] The presence of avidin can significantly increase the catalytic capacity of Rhodium(I) cofactor in aqueous phosphate buffer. Another pioneering work was conducted by Kaiseret al. where carboxypeptidase A (CPA) was repurposed into an oxidase by substituting Zn(II) center by Cu(II), for the oxidation of ascorbic acid.[9]
The real potential of ArMs was unleashed when recombinant protein production was developed, namely in 1997 Distefano and Davies reported a scaffold modification of a recombinant adipocyte lipid-binding protein (ALBP) with iodoacetamido-1,10-phenanthroline coordinating Cu(II) for the stereoselective hydrolysis of racemic esters.[10]
Formation
An artificial oxidase based on Cu(II)-bipyridine complex linked to the cysteine in the active site of adipocyte lipid binding protein (PDB: 1A18). Artificial ligand showed in red (copper not shown).
Abiotic cofactor anchoring
Four strategies have been used to assemble ArMs:[6]
Covalent immobilization of a metal-containing catalytic moiety by an irreversible reaction with the protein;
Supramolecular interactions between a protein and a high-affinity substrate could be used to anchor a metal cofactor;
The metal substitution in a natural metalloenzyme can result in a novel catalytic activity to the protein. The metal could be part of a prosthetic group (e.g., heme) or bound to amino acids;
Amino acids with Lewis-basic properties in a hydrophobic pocket could interact with coordinatively unsaturated metal center.
These four strategies led to a great progress in the field of artificial metalloenzymes since the beginning of the 21st century, unlocking exceptional selectivity for new-to-nature reactions.
Covalent
Different approaches to anchoring artificial metal cofactors. (Ball: Protein; Square: Metal cofactors)
With the development of bioconjugation technology, there are plenty of strategies to covalently bind an artificial metallocofactor onto a protein scaffold:
post-translational bioorthogonal modification based on Amber stop codon suppression (e.g., Click chemistry)[14]
enzyme active site modification (e.g., covalent bond formation between lipase and lipase inhibitor).[15]
Supramolecular
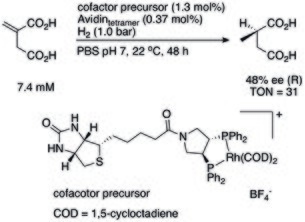
Streptavidin or avidin in combination with biotinylated artificial metal cofactors is the most commonly used supramolecular strategy to make ArMs.[16] In the early example from Ward et al. shown below, the ligand of Ru(I) complex was covalently linked to biotin and then the whole complex was anchored to streptavidin thanks to a specific and strong biotin-streptavidin interaction.[17] The formed ArM can catalyze the reduction of prochiralketones. Taking advantages of protein evolvability, different mutants of streptavidin can achieve different stereoselectivity. Throughout the years, many streptavidin-based enzymes were developed, enabling catalysis of very complex transformations in water, under ambient conditions.
An ArM using biotin-streptavidin interaction to anchor artificial metal cofactor (PDB: 2QCB)
Besides biotin-streptavidin based ArMs, another important example of using supramolecular iassembly strategy is antigen-antibody recognition. First reported in 1989 by Lerner et al.., a monoclonal antibody-based ArM is raised to hydrolyze specific peptide.[18]
Another interesting scaffold used as a platform for supramolecularly assembled ArMs are multidrug resistance regulators (MDRs), particularly a PadR family of proteins without native catalytic activity, whose function in nature is the recognition of foreign agents and to activate subsequent cellular response.[19] Among them, Lactococcal multidrug resistance regulator (LmrR) was mainly used to create ArMs, using different strategies, including the supramolecular one. Namely, Roelfes et al. incorporated Cu(II) phenanthroline complex in the hydrophobic pocket of LmrR and performed Friedel-Crafts reaction enantioselectively;[20][21] and Fe heme complex which catalyzed cyclopropanation enantioselectively.[22]
Metal substitution in a natural cofactor
Metal substitution
This strategy involves substitution of a native metal center in a metallocofactor, by another metal, that might or might not be already present in living systems.[23] In this way, electronic and steric properties of the catalytic active site are altered compared to the wild-type enzyme, and novel catalytic pathways are unlocked.
Dative
BpyAla and HQAla have been successfully incorporated in protein scaffolds and used to selectively coordinate different metals for various types of catalysis
The dative anchoring strategy uses natural amino acid residue in the protein scaffold like His, Cys, Glu, Asp and Ser to coordinate to a metal center. Like the first example of Pd-fibroin, dative anchoring to natural amino acids is not commonly used nowadays and often resulted in a more ambiguous binding site for metal compared with previous three methods.
However, these challenges can be overcome by in vivo incorporating metal-chelatingnon-canonical amino acids (ncAAs)[24] in the protein scaffold. These genetically encoded ncAAs' side chains have chelating moieties, such as 2,2'-bipyridine (3-(2,2'-bipyridin-5-yl)-L-alanine)[25] and 8-hydroxyquinoline (2-amino-3-(8-hydroxyquinolin-3-yl)propanoic acid)[26] that can selectively coordinate different metals. Combining protein scaffolds featuring chelating ncAAs with different metals yields exceptionally selective artificial metalloenzymes with various application potentials.[5] ncAAs are usually incorporated through the means of Amber stop codonsuppression, via the orthogonal translation system (OTS).[24]
Metal-chelating non-canonical amino acids can also be introduced to a protein scaffold in vitro, for example by covalently linking a bromine-substituted picolines to native cysteine residues. The cysteine-sulphur substitutes the bromine, forming a picoline-substituted cysteine which in turn can coordinate a metallocofactor.[27]
Natural Metalloenzymes repurposing
In addition to anchoring artificial metal center in the protein scaffold, researchers like Frances Arnold and Yang Yang focused on changing the native environment of natural metallocofactors. Due to the large sequence space that can be evolved in natural metalloenzymes, they can be evolved to catalyse non-native transformations. This process is known as enzyme repurposing. Directed evolution is commonly used to tailor the catalytic capacity and repurpose the enzyme function. Mostly based on native porphyrin-metallocofactor, Arnold's lab has developed many ArMs catalysing regioselective and/or enantioselective transformations, such as Carbon-Boron bond formation,[28]carbene insertion,[29] and aminohydroxylation[30] by evolving the sequence context of the corresponding ArMs.
↑ Leurs M, Dorn B, Wilhelm S, Manisegaran M, Tiller JC (July 2018). "Multicore Artificial Metalloenzymes Derived from Acylated Proteins as Catalysts for the Enantioselective Dihydroxylation and Epoxidation of Styrene Derivatives". Chemistry: A European Journal. 24 (42): 10859–10867. doi:10.1002/chem.201802185. PMID29808506.
↑ Wilson ME, Whitesides GM (January 1978). "Conversion of a protein to a homogeneous asymmetric hydrogenation catalyst by site-specific modification with a diphosphinerhodium(I) moiety". Journal of the American Chemical Society. 100 (1): 306–307. Bibcode:1978JAChS.100..306W. doi:10.1021/ja00469a064. ISSN0002-7863.
↑ Onoda A, Kihara Y, Fukumoto K, Sano Y, Hayashi T (August 2014). "Photoinduced Hydrogen Evolution Catalyzed by a Synthetic Diiron Dithiolate Complex Embedded within a Protein Matrix". ACS Catalysis. 4 (8): 2645–2648. doi:10.1021/cs500392e.
↑ Davies RR, Distefano A (December 1997). "A Semisynthetic Metalloenzyme Based on a Protein Cavity That Catalyzes the Enantioselective Hydrolysis of Ester and Amide Substrates". Journal of the American Chemical Society. 119 (48): 11643–11652. Bibcode:1997JAChS.11911643D. doi:10.1021/ja970820k. ISSN0002-7863.
↑ Platis IE, Ermácora MR, Fox RO (November 1993). "Oxidative polypeptide cleavage mediated by EDTA-Fe covalently linked to cysteine residues". Biochemistry. 32 (47): 12761–7. doi:10.1021/bi00210a027. PMID8251497.
↑ Kruithof CA, Casado MA, Guillena G, Egmond MR, van der Kerk-van Hoof A, Heck AJ, etal. (November 2005). "Lipase active-site-directed anchoring of organometallics: metallopincer/protein hybrids". Chemistry: A European Journal. 11 (23): 6869–77. doi:10.1002/chem.200500671. PMID16224766.
↑ Creus M, Pordea A, Rossel T, Sardo A, Letondor C, Ivanova A, etal. (2008). "X-ray structure and designed evolution of an artificial transfer hydrogenase". Angewandte Chemie. 47 (8): 1400–4. doi:10.1002/anie.200704865. PMID18176932.
↑ Yokoi N, Inaba H, Terauchi M, Stieg AZ, Sanghamitra NJ, Koshiyama T, etal. (September 2010). "Construction of robust bio-nanotubes using the controlled self-assembly of component proteins of bacteriophage T4". Small. 6 (17): 1873–9. doi:10.1002/smll.201000772. PMID20661999.
↑ Podtetenieff J, Taglieber A, Bill E, Reijerse EJ, Reetz MT (July 2010). "An artificial metalloenzyme: creation of a designed copper binding site in a thermostable protein". Angewandte Chemie. 49 (30): 5151–5. doi:10.1002/anie.201002106. PMID20572232.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.