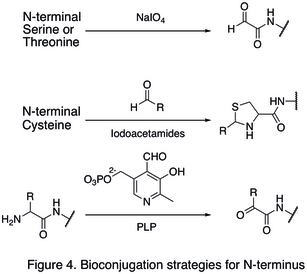
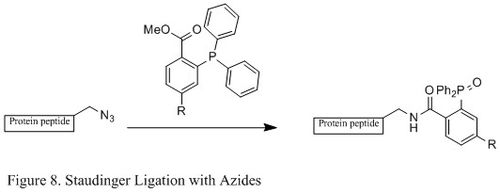
This article may be too technical for most readers to understand.(October 2022) |

Bioconjugation is a chemical strategy to form a stable covalent link between two molecules, at least one of which is a biomolecule. Methods to conjugate biomolecules are applied in various fields, including medicine, diagnostics, biocatalysis and materials. Synthetically modified biomolecules can have diverse functionalities, such as tracking cellular events, revealing enzyme function, determining protein biodistribution, imaging specific biomarkers, vaccination [1] and delivering drugs to targeted cells. [2] [3] [4] [5]
Contents
- Common Bioconjugation Reactions
- Reaction with Natural Amino Acids
- Bioorthogonal Reactions: On Unique Functional Groups
- Transition Metal-Mediated Bioconjugation Reactions
- Alkylation
- Arylation
- Examples of Applied Bioconjugation Techniques
- Growth Factors
- See also
- References
Bioconjugation is a crucial strategy that links these modified biomolecules with different substrates. Besides applications in biomedical research, bioconjugation has recently also gained importance in nanotechnology such as bioconjugated quantum dots.
The most common types of bioconjugation include coupling of a small molecule (such as biotin, a fluorescent dye, or a pharmaceutical drug) to a protein. Antibody-drug conjugates such as Brentuximab vedotin and Gemtuzumab ozogamicin are examples falling into this category. [6] Other less common molecules used in bioconjugation are oligosaccharides, nucleic acids, synthetic polymers such as polyethylene glycol, [7] and carbon nanotubes. [8] Protein-protein conjugations, such as the coupling of an antibody to an enzyme, or the linkage of protein complexes, are also facilitated via bioconjugations. [9] [10] [11]