
The proteome is the entire set of proteins that is, or can be, expressed by a genome, cell, tissue, or organism at a certain time. It is the set of expressed proteins in a given type of cell or organism, at a given time, under defined conditions. Proteomics is the study of the proteome.

Proteomics is the large-scale study of proteins. Proteins are vital parts of living organisms, with many functions such as the formation of structural fibers of muscle tissue, enzymatic digestion of food, or synthesis and replication of DNA. In addition, other kinds of proteins include antibodies that protect an organism from infection, and hormones that send important signals throughout the body.

Structural genomics seeks to describe the 3-dimensional structure of every protein encoded by a given genome. This genome-based approach allows for a high-throughput method of structure determination by a combination of experimental and modeling approaches. The principal difference between structural genomics and traditional structural prediction is that structural genomics attempts to determine the structure of every protein encoded by the genome, rather than focusing on one particular protein. With full-genome sequences available, structure prediction can be done more quickly through a combination of experimental and modeling approaches, especially because the availability of large number of sequenced genomes and previously solved protein structures allows scientists to model protein structure on the structures of previously solved homologs.
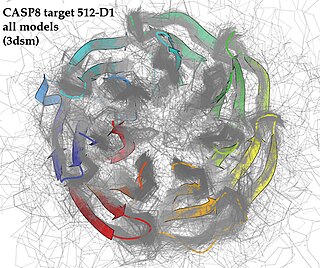
Critical Assessment of Structure Prediction (CASP), sometimes called Critical Assessment of Protein Structure Prediction, is a community-wide, worldwide experiment for protein structure prediction taking place every two years since 1994. CASP provides research groups with an opportunity to objectively test their structure prediction methods and delivers an independent assessment of the state of the art in protein structure modeling to the research community and software users. Even though the primary goal of CASP is to help advance the methods of identifying protein three-dimensional structure from its amino acid sequence many view the experiment more as a “world championship” in this field of science. More than 100 research groups from all over the world participate in CASP on a regular basis and it is not uncommon for entire groups to suspend their other research for months while they focus on getting their servers ready for the experiment and on performing the detailed predictions.
grid.org was a website and online community established in 2001 for cluster computing and grid computing software users. For six years it operated several different volunteer computing projects that allowed members to donate their spare computer cycles to worthwhile causes. In 2007, it became a community for open source cluster and grid computing software. After around 2010 it redirected to other sites.

In biology and other experimental sciences, an in silico experiment is one performed on computer or via computer simulation. The phrase is pseudo-Latin for 'in silicon', referring to silicon in computer chips. It was coined in 1987 as an allusion to the Latin phrases in vivo, in vitro, and in situ, which are commonly used in biology. The latter phrases refer, respectively, to experiments done in living organisms, outside living organisms, and where they are found in nature.
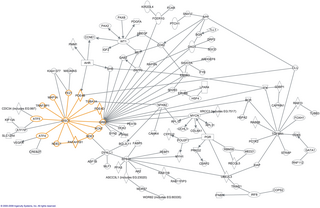
In molecular biology, an interactome is the whole set of molecular interactions in a particular cell. The term specifically refers to physical interactions among molecules but can also describe sets of indirect interactions among genes.

World Community Grid (WCG) is an effort to create the world's largest volunteer computing platform to tackle scientific research that benefits humanity. Launched on November 16, 2004, with proprietary Grid MP client from United Devices and adding support for Berkeley Open Infrastructure for Network Computing (BOINC) in 2005, World Community Grid eventually discontinued the Grid MP client and consolidated on the BOINC platform in 2008. In September 2021, it was announced that IBM transferred ownership to the Krembil Research Institute of University Health Network in Toronto, Ontario.
Rosetta is a software package for protein structure prediction. Originally introduced by the Baker laboratory at the University of Washington in 1998 as an ab initio approach to structure prediction, Rosetta has since branched into several development streams and distinct services, providing features such as macromolecular docking and protein design. Many of the graduate students and other researchers involved in Rosetta's initial development have since moved to other universities and research institutions, and subsequently enhanced different parts of the Rosetta project.
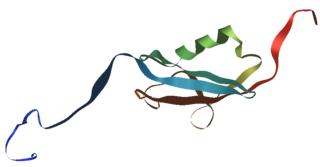
In molecular biology, SUMOproteins are a family of small proteins that are covalently attached to and detached from other proteins in cells to modify their function. This process is called SUMOylation. SUMOylation is a post-translational modification involved in various cellular processes, such as nuclear-cytosolic transport, transcriptional regulation, apoptosis, protein stability, response to stress, and progression through the cell cycle.
The contact order of a protein is a measure of the locality of the inter-amino acid contacts in the protein's native state tertiary structure. It is calculated as the average sequence distance between residues that form native contacts in the folded protein divided by the total length of the protein. Higher contact orders indicate longer folding times, and low contact order has been suggested as a predictor of potential downhill folding, or protein folding that occurs without a free energy barrier. This effect is thought to be due to the lower loss of conformational entropy associated with the formation of local as opposed to nonlocal contacts.

David Baker is an American biochemist and computational biologist who has pioneered methods to predict and design the three-dimensional structures of proteins. He is the Henrietta and Aubrey Davis Endowed Professor in Biochemistry and an adjunct professor of genome sciences, bioengineering, chemical engineering, computer science, and physics at the University of Washington. He serves as the director of the Rosetta Commons, a consortium of labs and researchers that develop biomolecular structure prediction and design software. The problem of protein structure prediction to which Baker has contributed significantly has now been largely solved by DeepMind using artificial intelligence. Baker is a Howard Hughes Medical Institute investigator and a member of the United States National Academy of Sciences. He is also the director of the University of Washington's Institute for Protein Design.
In computational biology, de novo protein structure prediction refers to an algorithmic process by which protein tertiary structure is predicted from its amino acid primary sequence. The problem itself has occupied leading scientists for decades while still remaining unsolved. According to Science, the problem remains one of the top 125 outstanding issues in modern science. At present, some of the most successful methods have a reasonable probability of predicting the folds of small, single-domain proteins within 1.5 angstroms over the entire structure.

Foldit is an online puzzle video game about protein folding. It is part of an experimental research project developed by the University of Washington, Center for Game Science, in collaboration with the UW Department of Biochemistry. The objective of Foldit is to fold the structures of selected proteins as perfectly as possible, using tools provided in the game. The highest scoring solutions are analyzed by researchers, who determine whether or not there is a native structural configuration that can be applied to relevant proteins in the real world. Scientists can then use these solutions to target and eradicate diseases and create biological innovations. A 2010 paper in the science journal Nature credited Foldit's 57,000 players with providing useful results that matched or outperformed algorithmically computed solutions.
Richard Bonneau is an American computational biologist and data scientist whose primary research is in the following areas: learning networks from functional genomics data, predicting and designing protein and peptiodomimetic structure and applying data science to social networks. A professor at New York University, he holds appointments in the department of biology, the Center for Data Science and the Courant Institute of Mathematical Sciences.
Décrypthon is a project which uses grid computing resources to contribute to medical research. The word is a portmanteau of the French word "décrypter" and "telethon".
Edward Marcotte is a professor of biochemistry at The University of Texas at Austin, working in genetics, proteomics, and bioinformatics. Marcotte is an example of a computational biologist who also relies on experiments to validate bioinformatics-based predictions.
The Human Proteome Project (HPP) is a collaborative effort coordinated by the Human Proteome Organization. Its stated goal is to experimentally observe all of the proteins produced by the sequences translated from the human genome.
The human interactome is the set of protein–protein interactions that occur in human cells. The sequencing of reference genomes, in particular the Human Genome Project, has revolutionized human genetics, molecular biology, and clinical medicine. Genome-wide association study results have led to the association of genes with most Mendelian disorders, and over 140 000 germline mutations have been associated with at least one genetic disease. However, it became apparent that inherent to these studies is an emphasis on clinical outcome rather than a comprehensive understanding of human disease; indeed to date the most significant contributions of GWAS have been restricted to the “low-hanging fruit” of direct single mutation disorders, prompting a systems biology approach to genomic analysis. The connection between genotype and phenotype remain elusive, especially in the context of multigenic complex traits and cancer. To assign functional context to genotypic changes, much of recent research efforts have been devoted to the mapping of the networks formed by interactions of cellular and genetic components in humans, as well as how these networks are altered by genetic and somatic disease.
Michael P. Snyder is an American genomicist who is the Stanford B. Ascherman Professor and as of 2009, chair of genetics and director of genomics and personalized medicine at Stanford University, and the former director of the Yale Center for Genomics and Proteomics. He was elected to the American Academy of Arts and Sciences in 2015. During his tenure as chair of the department at Stanford, U.S. News & World Report has ranked Stanford University first or tied for first in genetics, genomics and bioinformatics under his leadership.
