
Protein tertiary structure is the three dimensional shape of a protein. The tertiary structure will have a single polypeptide chain "backbone" with one or more protein secondary structures, the protein domains. Amino acid side chains may interact and bond in a number of ways. The interactions and bonds of side chains within a particular protein determine its tertiary structure. The protein tertiary structure is defined by its atomic coordinates. These coordinates may refer either to a protein domain or to the entire tertiary structure. A number of tertiary structures may fold into a quaternary structure.
Folding@home is a distributed computing project aimed to help scientists develop new therapeutics for a variety of diseases by the means of simulating protein dynamics. This includes the process of protein folding and the movements of proteins, and is reliant on simulations run on volunteers' personal computers. Folding@home is currently based at the University of Pennsylvania and led by Greg Bowman, a former student of Vijay Pande.

Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. The main objective of structural bioinformatics is the creation of new methods of analysing and manipulating biological macromolecular data in order to solve problems in biology and generate new knowledge.
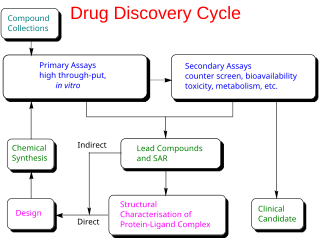
Drug design, often referred to as rational drug design or simply rational design, is the inventive process of finding new medications based on the knowledge of a biological target. The drug is most commonly an organic small molecule that activates or inhibits the function of a biomolecule such as a protein, which in turn results in a therapeutic benefit to the patient. In the most basic sense, drug design involves the design of molecules that are complementary in shape and charge to the biomolecular target with which they interact and therefore will bind to it. Drug design frequently but not necessarily relies on computer modeling techniques. This type of modeling is sometimes referred to as computer-aided drug design. Finally, drug design that relies on the knowledge of the three-dimensional structure of the biomolecular target is known as structure-based drug design. In addition to small molecules, biopharmaceuticals including peptides and especially therapeutic antibodies are an increasingly important class of drugs and computational methods for improving the affinity, selectivity, and stability of these protein-based therapeutics have also been developed.
grid.org was a website and online community established in 2001 for cluster computing and grid computing software users. For six years it operated several different volunteer computing projects that allowed members to donate their spare computer cycles to worthwhile causes. In 2007, it became a community for open source cluster and grid computing software. After around 2010 it redirected to other sites.

In biochemistry and pharmacology, a ligand is a substance that forms a complex with a biomolecule to serve a biological purpose. The etymology stems from Latin ligare, which means 'to bind'. In protein-ligand binding, the ligand is usually a molecule which produces a signal by binding to a site on a target protein. The binding typically results in a change of conformational isomerism (conformation) of the target protein. In DNA-ligand binding studies, the ligand can be a small molecule, ion, or protein which binds to the DNA double helix. The relationship between ligand and binding partner is a function of charge, hydrophobicity, and molecular structure.

In the field of molecular modeling, docking is a method which predicts the preferred orientation of one molecule to a second when a ligand and a target are bound to each other to form a stable complex. Knowledge of the preferred orientation in turn may be used to predict the strength of association or binding affinity between two molecules using, for example, scoring functions.

OpenEye Scientific Software is an American software company founded by Anthony Nicholls in 1997. It develops large-scale molecular modelling applications and toolkits. Following OpenEye's acquisition by Cadence Design Systems for $500 million in September 2022, the company was rebranded to OpenEye Cadence Molecular Sciences and operates as a business unit under Cadence.
Rosetta@home is a volunteer computing project researching protein structure prediction on the Berkeley Open Infrastructure for Network Computing (BOINC) platform, run by the Baker laboratory at the University of Washington. Rosetta@home aims to predict protein–protein docking and design new proteins with the help of about fifty-five thousand active volunteered computers processing at over 487,946 GigaFLOPS on average as of September 19, 2020. Foldit, a Rosetta@home videogame, aims to reach these goals with a crowdsourcing approach. Though much of the project is oriented toward basic research to improve the accuracy and robustness of proteomics methods, Rosetta@home also does applied research on malaria, Alzheimer's disease, and other pathologies.
Protein–ligand docking is a molecular modelling technique. The goal of protein–ligand docking is to predict the position and orientation of a ligand when it is bound to a protein receptor or enzyme. Pharmaceutical research employs docking techniques for a variety of purposes, most notably in the virtual screening of large databases of available chemicals in order to select likely drug candidates. There has been rapid development in computational ability to determine protein structure with programs such as AlphaFold, and the demand for the corresponding protein-ligand docking predictions is driving implementation of software that can find accurate models. Once the protein folding can be predicted accurately along with how the ligands of various structures will bind to the protein, the ability for drug development to progress at a much faster rate becomes possible.
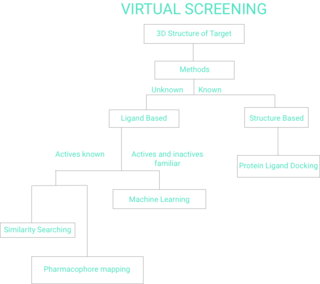
Virtual screening (VS) is a computational technique used in drug discovery to search libraries of small molecules in order to identify those structures which are most likely to bind to a drug target, typically a protein receptor or enzyme.
In computational biology, de novo protein structure prediction refers to an algorithmic process by which protein tertiary structure is predicted from its amino acid primary sequence. The problem itself has occupied leading scientists for decades while still remaining unsolved. According to Science, the problem remains one of the top 125 outstanding issues in modern science. At present, some of the most successful methods have a reasonable probability of predicting the folds of small, single-domain proteins within 1.5 angstroms over the entire structure.
In molecular modelling, docking is a method which predicts the preferred orientation of one molecule to another when bound together in a stable complex. In the case of protein docking, the search space consists of all possible orientations of the protein with respect to the ligand. Flexible docking in addition considers all possible conformations of the protein paired with all possible conformations of the ligand.

Smash Childhood Cancer is a World Community Grid volunteer computing subproject on the BOINC platform. It is based on World Community Grid's Help Childhood Cancer subproject which was a joint effort of Chiba University and the Chiba Cancer Center. Based on the results of that project, the Smash Childhood Cancer started in January 2017 looking for drug candidates targeting additional childhood cancers.

Help Cure Muscular Dystrophy is a volunteer computing project that runs on the BOINC platform.
Décrypthon is a project which uses grid computing resources to contribute to medical research. The word is a portmanteau of the French word "décrypter" and "telethon".
AutoDock is a molecular modeling simulation software. It is especially effective for protein-ligand docking. AutoDock 4 is available under the GNU General Public License. AutoDock is one of the most cited docking software applications in the research community. It is used by the FightAIDS@Home and OpenPandemics - COVID-19 projects run at World Community Grid, to search for antivirals against HIV/AIDS and COVID-19. In February 2007, a search of the ISI Citation Index showed more than 1,100 publications had been cited using the primary AutoDock method papers. As of 2009, this number surpassed 1,200.
The program UCSF DOCK was created in the 1980s by Irwin "Tack" Kuntz's Group, and was the first docking program. DOCK uses geometric algorithms to predict the binding modes of small molecules. Brian K. Shoichet, David A. Case, and Robert C.Rizzo are codevelopers of DOCK.

FightAIDS@Home is a volunteer computing project operated by the Olson Laboratory at The Scripps Research Institute. It runs on internet-connected home computers, and since July 2013 also runs on Android smartphones and tablets. It aims to use biomedical software simulation techniques to search for ways to cure or prevent the spread of HIV/AIDS.
LeDock is a proprietary flexible molecular docking software designed for the purpose of docking ligands with protein targets. It is available for Linux, macOS, and Windows.
