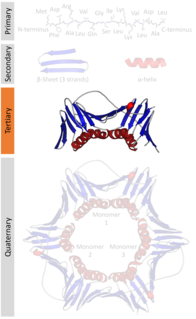
Protein tertiary structure is the three dimensional shape of a protein. The tertiary structure will have a single polypeptide chain "backbone" with one or more protein secondary structures, the protein domains. Amino acid side chains may interact and bond in a number of ways. The interactions and bonds of side chains within a particular protein determine its tertiary structure. The protein tertiary structure is defined by its atomic coordinates. These coordinates may refer either to a protein domain or to the entire tertiary structure. A number of tertiary structures may fold into a quaternary structure.
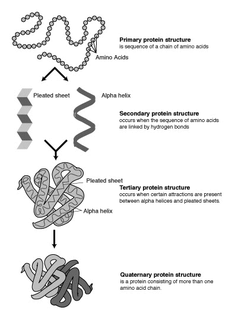
Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by computational biology; and it is important in medicine and biotechnology.

The Berkeley Open Infrastructure for Network Computing is an open-source middleware system for volunteer computing. Developed originally to support SETI@home, it became the platform for many other applications in areas as diverse as medicine, molecular biology, mathematics, linguistics, climatology, environmental science, and astrophysics, among others. The purpose of BOINC is to enable researchers to utilize processing resources of personal computers and other devices around the world.

Folding@home is a volunteer computing project aimed to help scientists develop new therapeutics for a variety of diseases by the means of simulating protein dynamics. This includes the process of protein folding and the movements of proteins, and is reliant on simulations run on volunteers' personal computers. Folding@home is currently based at the University of Pennsylvania and led by Greg Bowman, a former student of Vijay Pande.
Genome@home was a volunteer computing project run by Stefan Larson of Stanford University, and a sister project to Folding@home. Its goal was protein design and its applications, which had implications in many fields including medicine. Genome@home was run by the Pande Lab.
grid.org was a website and online community established in 2001 for cluster computing and grid computing software users. For six years it operated several different volunteer computing projects that allowed members to donate their spare computer cycles to worthwhile causes. In 2007, it became a community for open source cluster and grid computing software. After around 2010 it redirected to other sites.

World Community Grid (WCG) is an effort to create the world's largest volunteer computing platform to tackle scientific research that benefits humanity. Launched on November 16, 2004, with proprietary Grid MP client from United Devices and adding support for Berkeley Open Infrastructure for Network Computing (BOINC) in 2005, World Community Grid eventually discontinued the Grid MP client and consolidated on the BOINC platform in 2008. In September 2021, it was announced that IBM transferred ownership to the Krembil Research Institute of University Health Network in Toronto, Ontario.

The Human Proteome Folding Project (HPF) is a collaborative effort between New York University, the Institute for Systems Biology (ISB) and the University of Washington, using the Rosetta software developed by the Rosetta Commons.
Rosetta@home is a volunteer computing project researching protein structure prediction on the Berkeley Open Infrastructure for Network Computing (BOINC) platform, run by the Baker laboratory at the University of Washington. Rosetta@home aims to predict protein–protein docking and design new proteins with the help of about fifty-five thousand active volunteered computers processing at over 487,946 GigaFLOPS on average as of September 19, 2020. Foldit, a Rosetta@home videogame, aims to reach these goals with a crowdsourcing approach. Though much of the project is oriented toward basic research to improve the accuracy and robustness of proteomics methods, Rosetta@home also does applied research on malaria, Alzheimer's disease, and other pathologies.
Similarity Matrix of Proteins (SIMAP) is a database of protein similarities created using volunteer computing. It is freely accessible for scientific purposes. SIMAP uses the FASTA algorithm to precalculate protein similarity, while another application uses hidden Markov models to search for protein domains. SIMAP is a joint project of the Technical University of Munich, the Helmholtz Zentrum München, and the University of Vienna.

David Baker is an American biochemist and computational biologist who has pioneered methods to predict and design the three-dimensional structures of proteins. He is the Henrietta and Aubrey Davis Endowed Professor in Biochemistry and an adjunct professor of Genome Sciences, Bioengineering, Chemical Engineering, Computer Science, and Physics at the University of Washington. He serves as the Director of the Rosetta Commons, a consortium of labs and researchers that develop biomolecular structure prediction and design software. The problem of protein structure prediction to which Baker has contributed significantly has now been solved completely by DeepMind using artificial intelligence. Baker is a Howard Hughes Medical Institute investigator and a member of the United States National Academy of Sciences. He is also the director of the University of Washington's Institute for Protein Design.
In computational biology, de novo protein structure prediction refers to an algorithmic process by which protein tertiary structure is predicted from its amino acid primary sequence. The problem itself has occupied leading scientists for decades while still remaining unsolved. According to Science, the problem remains one of the top 125 outstanding issues in modern science. At present, some of the most successful methods have a reasonable probability of predicting the folds of small, single-domain proteins within 1.5 angstroms over the entire structure.

Volunteer computing is a type of distributed computing in which people donate their computers' unused resources to a research-oriented project, and sometimes in exchange for credit points. The fundamental idea behind it is that a modern desktop computer is sufficiently powerful to perform billions of operations a second, but for most users only between 10-15% of its capacity is used. Typical uses like basic word processing or web browsing leave the computer mostly idle.

proteins@home was a volunteer computing project that used the BOINC architecture. The project was run by the Department of Biology at École Polytechnique. The project began on December 28, 2006 and ended in June 2008.

Foldit is an online puzzle video game about protein folding. It is part of an experimental research project developed by the University of Washington, Center for Game Science, in collaboration with the UW Department of Biochemistry. The objective of Foldit is to fold the structures of selected proteins as perfectly as possible, using tools provided in the game. The highest scoring solutions are analyzed by researchers, who determine whether or not there is a native structural configuration that can be applied to relevant proteins in the real world. Scientists can then use these solutions to target and eradicate diseases and create biological innovations. A 2010 paper in the science journal Nature credited Foldit's 57,000 players with providing useful results that matched or outperformed algorithmically computed solutions.

Ibercivis was a volunteer computing platform which allows internet users to participate in scientific research by donating unused computer cycles to run scientific simulations and other tasks. The original project, which became operational in 2008, was a scientific collaboration between the Portuguese and Spanish governments, but it is open to the general public and scientific community, both within and beyond the Iberian Peninsula. The project's name is a portmanteau of Iberia and the Latin word civis, meaning 'citizen'.
GridRepublic is a BOINC Account Manager. It focuses on creating a clean and simple way to join and interact with BOINC. GridRepublic was started with a mission to raise public awareness and participation in volunteer computing with BOINC. GridRepublic was formed in 2004 by Matthew Blumberg as a mechanism to control the multiple projects from one place. The code for the BOINC software had to be redesigned to allow for the Account Manager system to be implemented.

POEM@Home was a volunteer computing project hosted by the Karlsruhe Institute of Technology and running on the Berkeley Open Infrastructure for Network Computing (BOINC) software platform. It modeled protein folding using Anfinsen's dogma. POEM@Home was started in 2007 and, due to advances using GPUs that rendered the BOINC program redundant, concluded in October 2016. The POEM@home applications were proprietary.
Charity Engine is a free PC app based on Berkeley University's BOINC software, run by The Worldwide Computer Company Limited. The project works by selling spare home computing power to universities and corporations, then sharing the profits between eight partner charities and periodic cash prize draws for the users; those running the Charity Engine BOINC software on their home computers. When there are no corporations purchasing the computing power, Charity Engine donates it to existing volunteer computing projects such as Rosetta@home, Einstein@Home, and Malaria Control, and prize draws are funded by donations.
