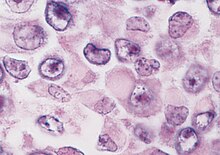
Wilms' tumor or Wilms tumor, also known as nephroblastoma, is a cancer of the kidneys that typically occurs in children, and occurs most commonly as a renal tumor in child patients. It is named after Max Wilms, the German surgeon (1867–1918) who first described it.

Oligodendrogliomas are a type of glioma that are believed to originate from the oligodendrocytes of the brain or from a glial precursor cell. They occur primarily in adults but are also found in children.
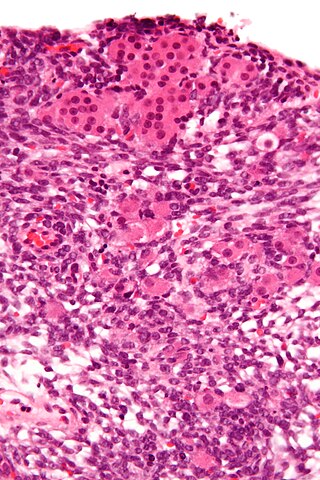
Sertoli–Leydig cell tumour is a group of tumors composed of variable proportions of Sertoli cells, Leydig cells, and in the case of intermediate and poorly differentiated neoplasms, primitive gonadal stroma and sometimes heterologous elements.

Fibrosarcoma is a malignant mesenchymal tumour derived from fibrous connective tissue and characterized by the presence of immature proliferating fibroblasts or undifferentiated anaplastic spindle cells in a storiform pattern. Fibrosarcomas mainly arise in people between the ages of 25 and 79. It originates in fibrous tissues of the bone and invades long or flat bones such as the femur, tibia, and mandible. It also involves the periosteum and overlying muscle.

Loss of heterozygosity (LOH) is a type of genetic abnormality in diploid organisms in which one copy of an entire gene and its surrounding chromosomal region are lost. Since diploid cells have two copies of their genes, one from each parent, a single copy of the lost gene still remains when this happens, but any heterozygosity is no longer present.
A blastoma is a type of cancer, more common in children, that is caused by malignancies in precursor cells, often called blasts. Examples are nephroblastoma, medulloblastoma, and retinoblastoma. The suffix -blastoma is used to imply a tumor of primitive, incompletely differentiated cells, e.g., chondroblastoma is composed of cells resembling the precursor of chondrocytes.
WAGR syndrome is a rare genetic syndrome in which affected children are predisposed to develop Wilms tumour, Aniridia, Genitourinary anomalies, and mental Retardation. The G is sometimes instead given as "gonadoblastoma," since the genitourinary anomalies can include tumours of the gonads.
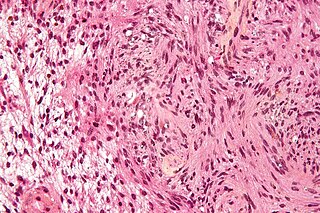
Schwannomatosis is an extremely rare genetic disorder closely related to the more-common disorder neurofibromatosis (NF). Originally described in Japanese patients, it consists of multiple cutaneous schwannomas, central nervous system tumors, and other neurological complications, excluding hallmark signs of NF. The exact frequency of schwannomatosis cases is unknown, although some populations have noted frequencies as few as 1 case per 1.7 million people.

22q13 deletion syndrome, also known as Phelan–McDermid syndrome (PMS), is a genetic disorder caused by deletions or rearrangements on the q terminal end of chromosome 22. Any abnormal genetic variation in the q13 region that presents with significant manifestations (phenotype) typical of a terminal deletion may be diagnosed as 22q13 deletion syndrome. There is disagreement among researchers as to the exact definition of 22q13 deletion syndrome. The Developmental Synaptopathies Consortium defines PMS as being caused by SHANK3 mutations, a definition that appears to exclude terminal deletions. The requirement to include SHANK3 in the definition is supported by many but not by those who first described 22q13 deletion syndrome.

Molecular cytogenetics combines two disciplines, molecular biology and cytogenetics, and involves the analysis of chromosome structure to help distinguish normal and cancer-causing cells. Human cytogenetics began in 1956 when it was discovered that normal human cells contain 46 chromosomes. However, the first microscopic observations of chromosomes were reported by Arnold, Flemming, and Hansemann in the late 1800s. Their work was ignored for decades until the actual chromosome number in humans was discovered as 46. In 1879, Arnold examined sarcoma and carcinoma cells having very large nuclei. Today, the study of molecular cytogenetics can be useful in diagnosing and treating various malignancies such as hematological malignancies, brain tumors, and other precursors of cancer. The field is overall focused on studying the evolution of chromosomes, more specifically the number, structure, function, and origin of chromosome abnormalities. It includes a series of techniques referred to as fluorescence in situ hybridization, or FISH, in which DNA probes are labeled with different colored fluorescent tags to visualize one or more specific regions of the genome. Introduced in the 1980s, FISH uses probes with complementary base sequences to locate the presence or absence of the specific DNA regions. FISH can either be performed as a direct approach to metaphase chromosomes or interphase nuclei. Alternatively, an indirect approach can be taken in which the entire genome can be assessed for copy number changes using virtual karyotyping. Virtual karyotypes are generated from arrays made of thousands to millions of probes, and computational tools are used to recreate the genome in silico.

An atypical teratoid rhabdoid tumor (AT/RT) is a rare tumor usually diagnosed in childhood. Although usually a brain tumor, AT/RT can occur anywhere in the central nervous system (CNS), including the spinal cord. About 60% will be in the posterior cranial fossa. One review estimated 52% in the posterior fossa, 39% are supratentorial primitive neuroectodermal tumors (sPNET), 5% are in the pineal, 2% are spinal, and 2% are multifocal.

Wilms tumor protein (WT33) is a protein that in humans is encoded by the WT1 gene on chromosome 11p.

SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1 is a protein that in humans is encoded by the SMARCB1 gene.

Congenital mesoblastic nephroma, while rare, is the most common kidney neoplasm diagnosed in the first three months of life and accounts for 3-5% of all childhood renal neoplasms. This neoplasm is generally non-aggressive and amenable to surgical removal. However, a readily identifiable subset of these kidney tumors has a more malignant potential and is capable of causing life-threatening metastases. Congenital mesoblastic nephroma was first named as such in 1967 but was recognized decades before this as fetal renal hamartoma or leiomyomatous renal hamartoma.

Epithelioid sarcoma is a rare soft tissue sarcoma arising from mesenchymal tissue and characterized by epithelioid-like features. It accounts for less than 1% of all soft tissue sarcomas. It was first definitively characterized by F.M. Enzinger in 1970. It commonly presents itself in the distal limbs of young adults as a small, soft mass or a cluster of bumps. A proximal version has also been described, frequently occurring in the upper extremities. Less commonly, cases are reported in the pelvis, vulva, penis, and spine.
Virtual karyotype is the digital information reflecting a karyotype, resulting from the analysis of short sequences of DNA from specific loci all over the genome, which are isolated and enumerated. It detects genomic copy number variations at a higher resolution for level than conventional karyotyping or chromosome-based comparative genomic hybridization (CGH). The main methods used for creating virtual karyotypes are array-comparative genomic hybridization and SNP arrays.

The WHOclassification of tumours of the central nervous system is a World Health Organization Blue Book that defines, describes and classifies tumours of the central nervous system (CNS).
Large cell lung carcinoma with rhabdoid phenotype (LCLC-RP) is a rare histological form of lung cancer, currently classified as a variant of large cell lung carcinoma (LCLC). In order for a LCLC to be subclassified as the rhabdoid phenotype variant, at least 10% of the malignant tumor cells must contain distinctive structures composed of tangled intermediate filaments that displace the cell nucleus outward toward the cell membrane. The whorled eosinophilic inclusions in LCLC-RP cells give it a microscopic resemblance to malignant cells found in rhabdomyosarcoma (RMS), a rare neoplasm arising from transformed skeletal muscle. Despite their microscopic similarities, LCLC-RP is not associated with rhabdomyosarcoma.
Pediatric ependymomas are similar in nature to the adult form of ependymoma in that they are thought to arise from radial glial cells lining the ventricular system. However, they differ from adult ependymomas in which genes and chromosomes are most often affected, the region of the brain they are most frequently found in, and the prognosis of the patients. Children with certain hereditary diseases, such as neurofibromatosis type II (NF2), have been found to be more frequently afflicted with this class of tumors, but a firm genetic link remains to be established. Symptoms associated with the development of pediatric ependymomas are varied, much like symptoms for a number of other pediatric brain tumors including vomiting, headache, irritability, lethargy, and changes in gait. Although younger children and children with invasive tumor types generally experience less favorable outcomes, total removal of the tumors is the most conspicuous prognostic factor for both survival and relapse.

22q11.2 distal deletion syndrome is a rare genetic condition caused by a tiny missing part of one of the body's 46 chromosomes – chromosome 22. 22q11.2 distal deletion syndrome appears to be a recurrent genomic disorder distinct from 22q11.2 deletion syndrome also known as DiGeorge syndrome and velocardiofacial syndrome.