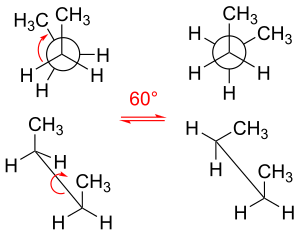
Various molecular structures formed only by rotation about single bonds
Rotation about single bond of butane to interconvert one conformation to another. The gauche conformation on the right is a conformer, while the eclipsed conformation on the left is a transition state between conformers. Above: Newman projection; below: depiction of spatial orientation.
In chemistry, rotamers are chemical species that differ from one another primarily due to rotations about one single bond. Various arrangements of atoms in a molecule that differ by rotation about single bonds can also be referred to as conformations. Conformations, which represent local minima on the potential energy surface, are called conformers.[1] Conformers can differ from one another due to rotation of multiple bonds; rotamers are a subset of conformers.[2] Conformers/rotamers usually differ little in their energies, so they are almost never separable in a practical sense. Rotations about single bonds are subject to small energy barriers.[3] When the time scale for interconversion is long enough for isolation of individual rotamers (usually arbitrarily defined as a half-life of interconversion of 1000 seconds or longer), the species are termed atropisomers.[4][5][6] The ring-flip of substituted cyclohexanes constitutes a common form of conformers.[7]
The study of the energetics of bond rotation is referred to as conformational analysis.[8] In some cases, conformational analysis can be used to predict and explain product selectivity, mechanisms, and rates of reactions.[9] Conformational analysis also plays an important role in rational, structure-based drug design.
rotamer: One of a set of conformers arising from restricted rotation about one single bond.[10]
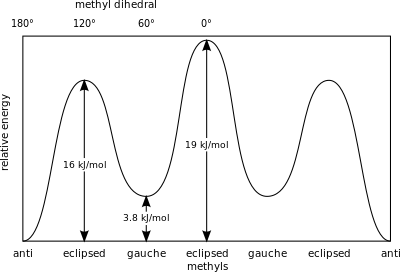
Relative conformation energy diagram of butane as a function of dihedral angle. A: antiperiplanar, anti or trans. B: synclinal or gauche. C: anticlinal or eclipsed. D: synperiplanar or cis.
Rotating their carbon–carbon bonds, the molecules ethane and propane have three local energy minima. They are structurally and energetically equivalent, and are called the staggered conformers. For each molecule, the three substituents emanating from each carbon–carbon bond are staggered, with each H–C–C–H dihedral angle (and H–C–C–CH3 dihedral angle in the case of propane) equal to 60° (or approximately equal to 60° in the case of propane). The three eclipsed conformations, in which the dihedral angles are zero, are transition states (energy maxima) connecting two equivalent energy minima, the staggered conformers. [citation needed]
The butane molecule is the simplest molecule for which single bond rotations result in two types of nonequivalent structures, known as the anti- and gauche-conformers (see figure).
For example, butane has three conformers relating to its two methyl (CH3) groups: two gauche conformers, which have the methyls ±60° apart and are enantiomeric, and an anti conformer, where the four carbon centres are coplanar and the substituents are 180° apart (refer to free energy diagram of butane). The energy separation between gauche and anti is 0.9kcal/mol associated with the strain energy of the gauche conformer. The anti conformer is, therefore, the most stable (≈0kcal/mol). The three eclipsed conformations with dihedral angles of 0°, 120°, and 240° are transition states between conformers.[8] Note that the two eclipsed conformations have distinct energies: at 0° the two methyl groups are eclipsed, resulting in higher energy (≈5kcal/mol) than at 120°, where the methyl groups are eclipsed with hydrogens (≈3.5kcal/mol).[12]
Mathematical analysis
A rough approximate function can illustrate the main features of the conformational analysis for unbranched linear alkanes with rotation around a central C–C bond (C1–C2 in ethane, C2–C3 in butane, C3–C4 in hexane, etc.).[13] The members of this series have the general formula C2nH4n+2 with the index n = 1, 2, 3, etc. It can be assumed that the angle strain is negligible in alkanes since the bond angles are all near the tetrahedral ideal. The energy profile is thus periodic with (120°) periodicity due to the threefold symmetry of sp3-hybridized carbon atoms. This suggests a sinusoidal potential energy function , typically modelled using a Fourier series truncated to the dominant terms:[14]
are coefficients representing the amplitude of the th harmonic, corresponding to various energy barriers due to torsional influences and asymmetry in steric interactions.
The factor of and the form ensure energy minima at staggered conformations and energy maxima at eclipsed conformations.
For alkanes, the dominant term is usually , reflecting the threefold rotational symmetry. Higher terms may be included for precision where steric effects vary. The primary contribution comes from torsional strain due to alkyl groups eclipsing, captured by the term. Steric interactions rise with the size of substituents (H– for ethane, CH3– for butane, C2H5– for hexane, etc.), taken into account by the term . The number of carbon atoms clearly influences the size of substituents on the central C–C bond. In general, for unbranched linear alkanes with even-numbered chains, there will be two Cn-1H2n-1 group substituents.
A parameterization using energy values derived from rotational spectroscopy data and theoretical calculations[15] gives the following simplified equation:
Here is given in kcal/mol and . This function largely neglects angle strain and long-range interactions for the members of the series.
Approximate potential function using a truncated Fourier series for the conformational analysis of unbranched linear alkanes with even-numbered chains.
While simple molecules can be described by these types of conformations, more complex molecules require the use of the Klyne–Prelog system to describe the conformers.[8]
More specific examples of conformations are detailed elsewhere:
Three isotherms are given in the diagram depicting the equilibrium distribution of two conformers at various temperatures. At a free energy difference of 0kcal/mol, this analysis gives an equilibrium constant of 1, meaning that two conformers exist in a 1:1 ratio. The two have equal free energy; neither is more stable, so neither predominates compared to the other. A negative difference in free energy means that a conformer interconverts to a thermodynamically more stable conformation, thus the equilibrium constant will always be greater than 1. For example, the ΔG° for the transformation of butane from the gauche conformer to the anti conformer is −0.47kcal/mol at 298 K.[18] This gives an equilibrium constant is about 2.2 in favor of the anti conformer, or a 31:69 mixture of gauche:anti conformers at equilibrium. Conversely, a positive difference in free energy means the conformer already is the more stable one, so the interconversion is an unfavorable equilibrium (K<1).
Population distribution of conformers
Boltzmann distribution% of lowest energy conformation in a two component equilibrating system at various temperatures (°C, color) and energy difference in kcal/mol (x-axis)
The left hand side is the proportion of conformer i in an equilibrating mixture of M conformers in thermodynamic equilibrium. On the right side, Ek (k = 1, 2, ..., M) is the energy of conformer k, R is the molar ideal gas constant (approximately equal to 8.314J/(mol·K) or 1.987 cal/(mol·K)), and T is the absolute temperature. The denominator of the right side is the partition function.
Factors contributing to the free energy of conformers
The effects of electrostatic and steric interactions of the substituents as well as orbital interactions such as hyperconjugation are responsible for the relative stability of conformers and their transition states. The contributions of these factors vary depending on the nature of the substituents and may either contribute positively or negatively to the energy barrier. Computational studies of small molecules such as ethane suggest that electrostatic effects make the greatest contribution to the energy barrier; however, the barrier is traditionally attributed primarily to steric interactions.[20][21]
Contributions to rotational energy barrier
In the case of cyclic systems, the steric effect and contribution to the free energy can be approximated by A values, which measure the energy difference when a substituent on cyclohexane in the axial as compared to the equatorial position. In large (>14 atom) rings, there are many accessible low-energy conformations which correspond to the strain-free diamond lattice.[22]
Observation of conformers
The short timescale of interconversion precludes the separation of conformer in most cases. Atropisomers are conformational isomers which can be separated due to restricted rotation.[23] The equilibrium between conformational isomers can be observed using a variety of spectroscopic techniques.[24]
Protein folding also generates conformers which can be observed. The Karplus equation relates the dihedral angle of vicinal protons to their J-coupling constants as measured by NMR. The equation aids in the elucidation of protein folding as well as the conformations of other rigid aliphatic molecules.[25] Protein side chains exhibit rotamers, whose distribution is determined by their steric interaction with different conformations of the backbone. This effect is evident from statistical analysis of the conformations of protein side chains in the Backbone-dependent rotamer library.[26]
Spectroscopy
Conformational dynamics can be monitored by variable temperature NMR spectroscopy. The technique applies to barriers of 8–14kcal/mol, and species exhibiting such dynamics are often called "fluxional". For example, in cyclohexane derivatives, the two chair conformers interconvert rapidly at room temperature. The ring-flip proceeds at a rates of approximately 105ring-flips/sec, with an overall energy barrier of 10 kcal/mol (42 kJ/mol). This barrier precludes separation at ambient temperatures.[27] However, at low temperatures below the coalescence point one can directly monitor the equilibrium by NMR spectroscopy and by dynamic, temperature dependent NMR spectroscopy the barrier interconversion.[28]
Besides NMR spectroscopy, IR spectroscopy is used to measure conformer ratios. For the axial and equatorial conformer of bromocyclohexane, νCBr differs by almost 50cm−1.[27]
Conformation-dependent reactions
Reaction rates are highly dependent on the conformation of the reactants. In many cases the dominant product arises from the reaction of the less prevalent conformer, by virtue of the Curtin-Hammett principle. This is typical for situations where the conformational equilibration is much faster than reaction to form the product. The dependence of a reaction on the stereochemical orientation is therefore usually only visible in configurational analysis, in which a particular conformation is locked by substituents. Prediction of rates of many reactions involving the transition between sp2 and sp3 states, such as ketone reduction, alcohol oxidation or nucleophilic substitution is possible if all conformers and their relative stability ruled by their strain is taken into account.[29]
One example where the rotamers become significant is elimination reactions, which involve the simultaneous removal of a proton and a leaving group from vicinal or antiperiplanar positions under the influence of a base.
Base-induced bimolecular dehydrohalogenation (an E2 type reaction mechanism). The optimum geometry for the transition state requires the breaking bonds to be antiperiplanar, as they are in the appropriate staggered conformation
The mechanism requires that the departing atoms or groups follow antiparallel trajectories. For open chain substrates this geometric prerequisite is met by at least one of the three staggered conformers. For some cyclic substrates such as cyclohexane, however, an antiparallel arrangement may not be attainable depending on the substituents which might set a conformational lock.[30] Adjacent substituents on a cyclohexane ring can achieve antiperiplanarity only when they occupy trans diaxial positions (that is, both are in axial position, one going up and one going down). [citation needed]
One consequence of this analysis is that trans-4-tert-butylcyclohexyl chloride cannot easily eliminate but instead undergoes substitution (see diagram below) because the most stable conformation has the bulky t-Bu group in the equatorial position, therefore the chloride group is not antiperiplanar with any vicinal hydrogen (it is gauche to all four). The thermodynamically unfavored conformation has the t-Bu group in the axial position, which is higher in energy by more than 5 kcal/mol (see A value).[31] As a result, the t-Bu group "locks" the ring in the conformation where it is in the equatorial position and substitution reaction is observed. On the other hand, cis-4-tert-butylcyclohexyl chloride undergoes elimination because antiperiplanarity of Cl and H can be achieved when the t-Bu group is in the favorable equatorial position.
Thermodynamically unfavored conformation of trans-4-tert-butylcyclohexyl chloride where the t-Bu group is in the axial position exerting 7-atom interactions.
The trans isomer can attain antiperiplanarity only via the unfavored axial conformer; therefore, it does not eliminate. The cis isomer is already in the correct geometry in its most stable conformation; therefore, it eliminates easily.
The repulsion between an axial t-butyl group and hydrogen atoms in the 1,3-diaxial position is so strong that the cyclohexane ring will revert to a twisted boat conformation. The strain in cyclic structures is usually characterized by deviations from ideal bond angles (Baeyer strain), ideal torsional angles (Pitzer strain) or transannular (Prelog) interactions.
Alkane stereochemistry
Alkane conformers arise from rotation around sp3 hybridised carbon–carbon sigma bonds. The smallest alkane with such a chemical bond, ethane, exists as an infinite number of conformations with respect to rotation around the C–C bond. Two of these are recognised as energy minimum (staggered conformation) and energy maximum (eclipsed conformation) forms. The existence of specific conformations is due to hindered rotation around sigma bonds, although a role for hyperconjugation is proposed by a competing theory. [citation needed]
The importance of energy minima and energy maxima is seen by extension of these concepts to more complex molecules for which stable conformations may be predicted as minimum-energy forms. The determination of stable conformations has also played a large role in the establishment of the concept of asymmetric induction and the ability to predict the stereochemistry of reactions controlled by steric effects. [citation needed]
In the example of staggered ethane in Newman projection, a hydrogen atom on one carbon atom has a 60° torsional angle or torsion angle[32] with respect to the nearest hydrogen atom on the other carbon so that steric hindrance is minimised. The staggered conformation is more stable by 12.5 kJ/mol than the eclipsed conformation, which is the energy maximum for ethane. In the eclipsed conformation the torsional angle is minimised.
staggered conformation left, eclipsed conformation right in Newman projection
In butane, the two staggered conformations are no longer equivalent and represent two distinct conformers:the anti-conformation (left-most, below) and the gauche conformation (right-most, below).
anti vs gauche conformations
Both conformations are free of torsional strain, but, in the gauche conformation, the two methyl groups are in closer proximity than the sum of their van der Waals radii. The interaction between the two methyl groups is repulsive (van der Waals strain), and an energy barrier results.
A measure of the potential energy stored in butane conformers with greater steric hindrance than the 'anti'-conformer ground state is given by these values:[33]
Relative energies of conformations of butane with respect to rotation of the central C-C bond.
The textbook explanation for the existence of the energy maximum for an eclipsed conformation in ethane is steric hindrance, but, with a C-C bond length of 154 pm and a Van der Waals radius for hydrogen of 120 pm, the hydrogen atoms in ethane are never in each other's way. The question of whether steric hindrance is responsible for the eclipsed energy maximum is a topic of debate to this day. One alternative to the steric hindrance explanation is based on hyperconjugation as analyzed within the Natural Bond Orbital framework.[34][35][36] In the staggered conformation, one C-H sigmabonding orbital donates electron density to the antibonding orbital of the other C-H bond. The energetic stabilization of this effect is maximized when the two orbitals have maximal overlap, occurring in the staggered conformation. There is no overlap in the eclipsed conformation, leading to a disfavored energy maximum. On the other hand, an analysis within quantitative molecular orbital theory shows that 2-orbital-4-electron (steric) repulsions are dominant over hyperconjugation.[37] A valence bond theory study also emphasizes the importance of steric effects.[38]
Nomenclature
Naming alkanes per standards listed in the IUPAC Gold Book is done according to the Klyne–Prelog system for specifying angles (called either torsional or dihedral angles) between substituents around a single bond:[32]
syn/anti peri/clinal
a torsion angle between 0° and ±90° is called syn (s)
a torsion angle between ±90° and 180° is called anti (a)
a torsion angle between 30° and 150° or between −30° and −150° is called clinal (c)
a torsion angle between 0° and ±30° or ±150° and 180° is called periplanar (p)
a torsion angle between 0° and ±30° is called synperiplanar (sp), also called syn- or cis- conformation
a torsion angle between 30° to 90° and −30° to −90° is called synclinal (sc), also called gauche or skew[39]
a torsion angle between 90° and 150° or −90° and −150° is called anticlinal (ac)
a torsion angle between ±150° and 180° is called antiperiplanar (ap), also called anti- or trans- conformation
Torsional strain or "Pitzer strain" refers to resistance to twisting about a bond.
↑ Ōki, Michinori (1983) Recent Advances in Atropisomerism, in Topics in Stereochemistry, Vol. 14 (N. L. Allinger, E. L. Eliel and S. H. Wilen, Eds.), Hoboken, NJ:John Wiley & Sons, pp. 1–82; published online in 2007, DOI: 10.1002/9780470147238.ch1, see and [permanent dead link], accessed 12 June 2014.
↑ Alkorta, Ibon; Jose Elguero; Christian Roussel; Nicolas Vanthuyne; Patrick Piras (2012). Atropisomerism and Axial Chirality in Heteroaromatic Compounds. Advances in Heterocyclic Chemistry. Vol.105. pp.1–188. doi:10.1016/B978-0-12-396530-1.00001-2. hdl:10261/62060. ISBN9780123965301.
↑ Hunt, Ian. "Stereochemistry". University of Calgary. Retrieved 28 October 2013.
↑ The standard enthalpy change ΔH° from gauche to anti is –0.88 kcal/mol. However, because there are two possible gauche forms, there is a statistical factor that needs to be taken into account as an entropic term. Thus, ΔG° = ΔH° – TΔS° = ΔH° + RT ln 2 = –0.88 kcal/mol + 0.41 kcal/mol = –0.47 kcal/mol, at 298 K.
1 2 Eliel, E. L.; Wilen, S. H.; Mander, L. N. (1994). Stereochemistry Of Organic Compounds. J. Wiley and Sons. ISBN978-0-471-01670-0.
↑ Jensen, Frederick R.; Bushweller, C. Hackett (1969-06-01). "Separation of conformers. II. Axial and equatorial isomers of chlorocyclohexane and trideuteriomethoxycyclohexane". Journal of the American Chemical Society. 91 (12): 3223–3225. Bibcode:1969JAChS..91.3223J. doi:10.1021/ja01040a022. ISSN0002-7863.
↑ Mo, Yirong; Wu, Wei; Song, Lingchun; Lin, Menghai; Zhang, Qianer; Gao, Jiali (2004-03-30). "The Magnitude of Hyperconjugation in Ethane: A Perspective from Ab Initio Valence Bond Theory". Angewandte Chemie International Edition. 43 (15). Wiley: 1986–1990. Bibcode:2004ACIE...43.1986M. doi:10.1002/anie.200352931. ISSN1433-7851. PMID15065281.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.