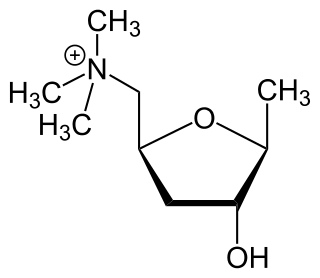
Muscarine, L-(+)-muscarine, or muscarin is a natural product found in certain mushrooms, particularly in Inocybe and Clitocybe species, such as the deadly C. dealbata. Mushrooms in the genera Entoloma and Mycena have also been found to contain levels of muscarine which can be dangerous if ingested. Muscarine has been found in harmless trace amounts in Boletus, Hygrocybe, Lactarius and Russula. Trace concentrations of muscarine are also found in Amanita muscaria, though the pharmacologically more relevant compound from this mushroom is the Z-drug-like alkaloid muscimol. A. muscaria fruitbodies contain a variable dose of muscarine, usually around 0.0003% fresh weight. This is very low and toxicity symptoms occur very rarely. Inocybe and Clitocybe contain muscarine concentrations up to 1.6%.

N-methyl-D-aspartic acid or N-methyl-D-aspartate (NMDA) is an amino acid derivative that acts as a specific agonist at the NMDA receptor mimicking the action of glutamate, the neurotransmitter which normally acts at that receptor. Unlike glutamate, NMDA only binds to and regulates the NMDA receptor and has no effect on other glutamate receptors. NMDA receptors are particularly important when they become overactive during, for example, withdrawal from alcohol as this causes symptoms such as agitation and, sometimes, epileptiform seizures.

Muscimol is one of the principal psychoactive constituents of Amanita muscaria and related species of mushroom. Muscimol is a potent and selective orthosteric agonist for the GABAA receptor and displays sedative-hypnotic, depressant and hallucinogenic psychoactivity. This colorless or white solid is classified as an isoxazole.

Epibatidine is a chlorinated alkaloid that is secreted by the Ecuadoran frog Epipedobates anthonyi and poison dart frogs from the Ameerega genus. It was discovered by John W. Daly in 1974, but its structure was not fully elucidated until 1992. Whether epibatidine is the first observed example of a chlorinated alkaloid remains controversial, due to challenges in conclusively identifying the compound from the limited samples collected by Daly. By the time that high-resolution spectrometry was used in 1991, there remained less than one milligram of extract from Daly's samples, raising concerns about possible contamination. Samples from other batches of the same species of frog failed to yield epibatidine.

The Pauson–Khand (PK) reaction is a chemical reaction, described as a [2+2+1] cycloaddition. In it, an alkyne, an alkene and carbon monoxide combine into a α,β-cyclopentenone in the presence of a metal-carbonyl catalyst.

Kainic acid, or kainate, is an acid that naturally occurs in some seaweed. Kainic acid is a potent neuroexcitatory amino acid agonist that acts by activating receptors for glutamate, the principal excitatory neurotransmitter in the central nervous system. Glutamate is produced by the cell's metabolic processes and there are four major classifications of glutamate receptors: NMDA receptors, AMPA receptors, kainate receptors, and the metabotropic glutamate receptors. Kainic acid is an agonist for kainate receptors, a type of ionotropic glutamate receptor. Kainate receptors likely control a sodium channel that produces excitatory postsynaptic potentials (EPSPs) when glutamate binds.

Orellanine or orellanin is a mycotoxin found in a group of mushrooms known as the Orellani of the family Cortinariaceae. Structurally, it is a bipyridine N-oxide compound somewhat related to the herbicide diquat.

Allodynia is a condition in which pain is caused by a stimulus that does not normally elicit pain. For example, bad sunburn can cause temporary allodynia, and touching sunburned skin, or running cold or warm water over it, can be very painful. It is different from hyperalgesia, an exaggerated response from a normally painful stimulus. The term comes from Ancient Greek άλλος (állos) 'other', and οδύνη (odúnē) 'pain'.

Di-tert-butyl dicarbonate is a reagent widely used in organic synthesis. Since this compound can be regarded formally as the acid anhydride derived from a tert-butoxycarbonyl (Boc) group, it is commonly referred to as Boc anhydride. This pyrocarbonate reacts with amines to give N-tert-butoxycarbonyl or so-called Boc derivatives. These carbamate derivatives do not behave as amines, which allows certain subsequent transformations to occur that would be incompatible with the amine functional group. The Boc group can later be removed from the amine using moderately strong acids. Thus, Boc serves as a protective group, for instance in solid phase peptide synthesis. Boc-protected amines are unreactive to most bases and nucleophiles, allowing for the use of the fluorenylmethyloxycarbonyl group (Fmoc) as an orthogonal protecting group.

Quisqualic acid is an agonist of the AMPA, kainate, and group I metabotropic glutamate receptors. It is one of the most potent AMPA receptor agonists known. It causes excitotoxicity and is used in neuroscience to selectively destroy neurons in the brain or spinal cord. Quisqualic acid occurs naturally in the seeds of Quisqualis species.

Seletracetam is a pyrrolidone-derived drug of the racetam family that is structurally related to levetiracetam. It was under development by UCB Pharmaceuticals as a more potent and effective anticonvulsant drug to replace levetiracetam but its development has been halted.

Apamin is an 18 amino acid globular peptide neurotoxin found in apitoxin (bee venom). Dry bee venom consists of 2–3% of apamin. Apamin selectively blocks SK channels, a type of Ca2+-activated K+ channel expressed in the central nervous system. Toxicity is caused by only a few amino acids, in particular cysteine1, lysine4, arginine13, arginine14 and histidine18. These amino acids are involved in the binding of apamin to the Ca2+-activated K+ channel. Due to its specificity for SK channels, apamin is used as a drug in biomedical research to study the electrical properties of SK channels and their role in the afterhyperpolarizations occurring immediately following an action potential.
The Fukuyama coupling is a coupling reaction taking place between a thioester and an organozinc halide in the presence of a palladium catalyst. The reaction product is a ketone. This reaction was discovered by Tohru Fukuyama et al. in 1998.

The Achmatowicz reaction, also known as the Achmatowicz rearrangement, is an organic synthesis in which a furan is converted to a dihydropyran. In the original publication by the Polish Chemist Osman Achmatowicz Jr. in 1971 furfuryl alcohol is reacted with bromine in methanol to 2,5-dimethoxy-2,5-dihydrofuran which rearranges to the dihydropyran with dilute sulfuric acid. Additional reaction steps, alcohol protection with methyl orthoformate and boron trifluoride) and then ketone reduction with sodium borohydride produce an intermediate from which many monosaccharides can be synthesised.
A convulsant is a drug which induces convulsions and/or epileptic seizures, the opposite of an anticonvulsant. These drugs generally act as stimulants at low doses, but are not used for this purpose due to the risk of convulsions and consequent excitotoxicity. Most convulsants are antagonists at either the GABAA or glycine receptors, or ionotropic glutamate receptor agonists. Many other drugs may cause convulsions as a side effect at high doses but only drugs whose primary action is to cause convulsions are known as convulsants. Nerve agents such as sarin, which were developed as chemical weapons, produce convulsions as a major part of their toxidrome, but also produce a number of other effects in the body and are usually classified separately. Dieldrin which was developed as an insecticide blocks chloride influx into the neurons causing hyperexcitability of the CNS and convulsions. The Irwin observation test and other studies that record clinical signs are used to test the potential for a drug to induce convulsions. Camphor, and other terpenes given to children with colds can act as convulsants in children who have had febrile seizures.

Tutin is a poisonous plant derivative found in New Zealand tutu plants. It acts as a potent antagonist of the glycine receptor, and has powerful convulsant effects. It is used in scientific research into the glycine receptor. It is sometimes associated with outbreaks of toxic honey poisoning when bees feed on honeydew exudate from the sap-sucking passion vine hopper insect, when the vine hoppers have been feeding on the sap of tutu bushes. Toxic honey is a rare event and is more likely to occur when comb honey is eaten directly from a hive that has been harvesting honeydew from passionvine hoppers feeding on tutu plants.
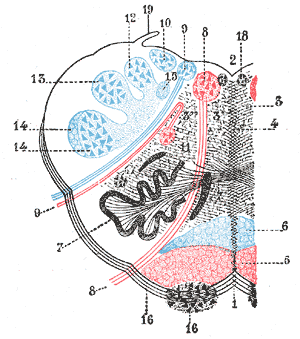
The rostral ventromedial medulla (RVM), or ventromedial nucleus of the spinal cord, is a group of neurons located close to the midline on the floor of the medulla oblongata. The rostral ventromedial medulla sends descending inhibitory and excitatory fibers to the dorsal horn spinal cord neurons. There are 3 categories of neurons in the RVM: on-cells, off-cells, and neutral cells. They are characterized by their response to nociceptive input. Off-cells show a transitory decrease in firing rate right before a nociceptive reflex, and are theorized to be inhibitory. Activation of off-cells, either by morphine or by any other means, results in antinociception. On-cells show a burst of activity immediately preceding nociceptive input, and are theorized to be contributing to the excitatory drive. Neutral cells show no response to nociceptive input.

Gelsemine (C20H22N2O2) is an indole alkaloid isolated from flowering plants of the genus Gelsemium, a plant native to the subtropical and tropical Americas, and southeast Asia, and is a highly toxic compound that acts as a paralytic, exposure to which can result in death. It has generally potent activity as an agonist of the mammalian glycine receptor, the activation of which leads to an inhibitory postsynaptic potential in neurons following chloride ion influx, and systemically, to muscle relaxation of varying intensity and deleterious effect. Despite its danger and toxicity, recent pharmacological research has suggested that the biological activities of this compound may offer opportunities for developing treatments related to xenobiotic or diet-induced oxidative stress, and of anxiety and other conditions, with ongoing research including attempts to identify safer derivatives and analogs to make use of gelsemine's beneficial effects.

Willardiine (correctly spelled with two successive i's) or (S)-1-(2-amino-2-carboxyethyl)pyrimidine-2,4-dione is a chemical compound that occurs naturally in the seeds of Mariosousa willardiana and Acacia sensu lato. The seedlings of these plants contain enzymes capable of complex chemical substitutions that result in the formation of free amino acids (See:#Synthesis). Willardiine is frequently studied for its function in higher level plants. Additionally, many derivates of willardiine are researched for their potential in pharmaceutical development. Willardiine was first discovered in 1959 by R. Gmelin, when he isolated several free, non-protein amino acids from Acacia willardiana (another name for Mariosousa willardiana) when he was studying how these families of plants synthesize uracilyalanines. A related compound, Isowillardiine, was concurrently isolated by a different group, and it was discovered that the two compounds had different structural and functional properties. Subsequent research on willardiine has focused on the functional significance of different substitutions at the nitrogen group and the development of analogs of willardiine with different pharmacokinetic properties. In general, Willardiine is the one of the first compounds studied in which slight changes to molecular structure result in compounds with significantly different pharmacokinetic properties.

Hafnium(IV) triflate or hafnium trifluoromethansulfonate is a salt with the formula Hf(OSO2CF3)4, also written as Hf(OTf)4. Hafnium triflate is used as an impure mixture as a catalyst. Hafnium (IV) has an ionic radius of intermediate range (Al < Ti < Hf < Zr < Sc < Ln) and has an oxophilic hard character typical of group IV metals. This solid is a stronger Lewis acid than its typical precursor hafnium tetrachloride, HfCl4, because of the strong electron-withdrawing nature of the four triflate groups, which makes it a great Lewis acid and has many uses including as a great catalyst at low Lewis acid loadings for electrophilic aromatic substitution and nucleophilic substitution reactions.
