
N-methyl-D-aspartic acid or N-methyl-D-aspartate (NMDA) is an amino acid derivative that acts as a specific agonist at the NMDA receptor mimicking the action of glutamate, the neurotransmitter which normally acts at that receptor. Unlike glutamate, NMDA only binds to and regulates the NMDA receptor and has no effect on other glutamate receptors. NMDA receptors are particularly important when they become overactive during, for example, withdrawal from alcohol as this causes symptoms such as agitation and, sometimes, epileptiform seizures.

The α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (also known as AMPA receptor, AMPAR, or quisqualate receptor) is an ionotropic transmembrane receptor for glutamate (iGluR) and predominantly Na+ ion channel that mediates fast synaptic transmission in the central nervous system (CNS). It has been traditionally classified as a non-NMDA-type receptor, along with the kainate receptor. Its name is derived from its ability to be activated by the artificial glutamate analog AMPA. The receptor was first named the "quisqualate receptor" by Watkins and colleagues after a naturally occurring agonist quisqualate and was only later given the label "AMPA receptor" after the selective agonist developed by Tage Honore and colleagues at the Royal Danish School of Pharmacy in Copenhagen. The GRIA2-encoded AMPA receptor ligand binding core (GluA2 LBD) was the first glutamate receptor ion channel domain to be crystallized.
An inhibitory postsynaptic potential (IPSP) is a kind of synaptic potential that makes a postsynaptic neuron less likely to generate an action potential. The opposite of an inhibitory postsynaptic potential is an excitatory postsynaptic potential (EPSP), which is a synaptic potential that makes a postsynaptic neuron more likely to generate an action potential. IPSPs can take place at all chemical synapses, which use the secretion of neurotransmitters to create cell-to-cell signalling. EPSPs and IPSPs compete with each other at numerous synapses of a neuron. This determines whether an action potential occurring at the presynaptic terminal produces an action potential at the postsynaptic membrane. Some common neurotransmitters involved in IPSPs are GABA and glycine.

An excitatory synapse is a synapse in which an action potential in a presynaptic neuron increases the probability of an action potential occurring in a postsynaptic cell. Neurons form networks through which nerve impulses travels, each neuron often making numerous connections with other cells of neurons. These electrical signals may be excitatory or inhibitory, and, if the total of excitatory influences exceeds that of the inhibitory influences, the neuron will generate a new action potential at its axon hillock, thus transmitting the information to yet another cell.

In excitotoxicity, nerve cells suffer damage or death when the levels of otherwise necessary and safe neurotransmitters such as glutamate become pathologically high, resulting in excessive stimulation of receptors. For example, when glutamate receptors such as the NMDA receptor or AMPA receptor encounter excessive levels of the excitatory neurotransmitter, glutamate, significant neuronal damage might ensue. Excess glutamate allows high levels of calcium ions (Ca2+) to enter the cell. Ca2+ influx into cells activates a number of enzymes, including phospholipases, endonucleases, and proteases such as calpain. These enzymes go on to damage cell structures such as components of the cytoskeleton, membrane, and DNA. In evolved, complex adaptive systems such as biological life it must be understood that mechanisms are rarely, if ever, simplistically direct. For example, NMDA in subtoxic amounts induces neuronal survival of otherwise toxic levels of glutamate.

Ligand-gated ion channels (LICs, LGIC), also commonly referred to as ionotropic receptors, are a group of transmembrane ion-channel proteins which open to allow ions such as Na+, K+, Ca2+, and/or Cl− to pass through the membrane in response to the binding of a chemical messenger (i.e. a ligand), such as a neurotransmitter.
Molecular neuroscience is a branch of neuroscience that observes concepts in molecular biology applied to the nervous systems of animals. The scope of this subject covers topics such as molecular neuroanatomy, mechanisms of molecular signaling in the nervous system, the effects of genetics and epigenetics on neuronal development, and the molecular basis for neuroplasticity and neurodegenerative diseases. As with molecular biology, molecular neuroscience is a relatively new field that is considerably dynamic.

Kainate receptors, or kainic acid receptors (KARs), are ionotropic receptors that respond to the neurotransmitter glutamate. They were first identified as a distinct receptor type through their selective activation by the agonist kainate, a drug first isolated from the algae Digenea simplex. They have been traditionally classified as a non-NMDA-type receptor, along with the AMPA receptor. KARs are less understood than AMPA and NMDA receptors, the other ionotropic glutamate receptors. Postsynaptic kainate receptors are involved in excitatory neurotransmission. Presynaptic kainate receptors have been implicated in inhibitory neurotransmission by modulating release of the inhibitory neurotransmitter GABA through a presynaptic mechanism.
Kainic acid, or kainate, is an acid that naturally occurs in some seaweed. Kainic acid is a potent neuroexcitatory amino acid agonist that acts by activating receptors for glutamate, the principal excitatory neurotransmitter in the central nervous system. Glutamate is produced by the cell's metabolic processes and there are four major classifications of glutamate receptors: NMDA receptors, AMPA receptors, kainate receptors, and the metabotropic glutamate receptors. Kainic acid is an agonist for kainate receptors, a type of ionotropic glutamate receptor. Kainate receptors likely control a sodium channel that produces excitatory postsynaptic potentials (EPSPs) when glutamate binds.

The metabotropic glutamate receptors, or mGluRs, are a type of glutamate receptor that are active through an indirect metabotropic process. They are members of the group C family of G-protein-coupled receptors, or GPCRs. Like all glutamate receptors, mGluRs bind with glutamate, an amino acid that functions as an excitatory neurotransmitter.

Glutamate receptors are synaptic and non synaptic receptors located primarily on the membranes of neuronal and glial cells. Glutamate is abundant in the human body, but particularly in the nervous system and especially prominent in the human brain where it is the body's most prominent neurotransmitter, the brain's main excitatory neurotransmitter, and also the precursor for GABA, the brain's main inhibitory neurotransmitter. Glutamate receptors are responsible for the glutamate-mediated postsynaptic excitation of neural cells, and are important for neural communication, memory formation, learning, and regulation.
Glutamate transporters are a family of neurotransmitter transporter proteins that move glutamate – the principal excitatory neurotransmitter – across a membrane. The family of glutamate transporters is composed of two primary subclasses: the excitatory amino acid transporter (EAAT) family and vesicular glutamate transporter (VGLUT) family. In the brain, EAATs remove glutamate from the synaptic cleft and extrasynaptic sites via glutamate reuptake into glial cells and neurons, while VGLUTs move glutamate from the cell cytoplasm into synaptic vesicles. Glutamate transporters also transport aspartate and are present in virtually all peripheral tissues, including the heart, liver, testes, and bone. They exhibit stereoselectivity for L-glutamate but transport both L-aspartate and D-aspartate.

DNQX (6,7-dinitroquinoxaline-2,3-dione) is a competitive antagonist at AMPA and kainate receptors, two ionotropic glutamate receptor (iGluR) subfamilies. It is used in a variety of molecular biology subfields, notably neurophysiology, to assist researchers in determining the properties of various types of ion channels and their potential applications in medicine.

Glutamate ionotropic receptor AMPA type subunit 2 is a protein that in humans is encoded by the GRIA2 gene and it is a subunit found in the AMPA receptors.

Glutamate receptor, ionotropic, kainate 1, also known as GRIK1, is a protein that in humans is encoded by the GRIK1 gene.
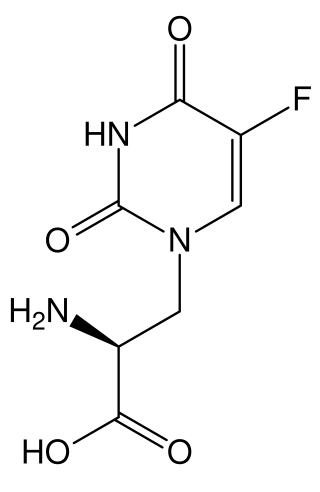
5-Fluorowillardiine is a selective agonist for the AMPA receptor, with only limited effects at the kainate receptor. It is an excitotoxic neurotoxin when used in vivo and so is rarely used in intact animals, but it is widely used to selectively stimulate AMPA receptors in vitro. It is structurally similar to the compound willardiine, which is also an agonist for the AMPA and kainate receptors. Willardiine occurs naturally in Mariosousa willardiana and Acacia sensu lato.

5-Iodowillardiine is a selective agonist for the kainate receptor, with only limited effects at the AMPA receptor. It is selective for kainate receptors composed of GluR5 subunits. It is an excitotoxic neurotoxin in vivo, but has proved highly useful for characterising the subtypes and function of the various kainate receptors in the brain and spinal cord.

Quisqualamine is the α-decarboxylated analogue of quisqualic acid, as well as a relative of the neurotransmitters glutamate and γ-aminobutyric acid (GABA). α-Decarboxylation of excitatory amino acids can produce derivatives with inhibitory effects. Indeed, unlike quisqualic acid, quisqualamine has central depressant and neuroprotective properties and appears to act predominantly as an agonist of the GABAA receptor and also to a lesser extent as an agonist of the glycine receptor, due to the facts that its actions are inhibited in vitro by GABAA antagonists like bicuculline and picrotoxin and by the glycine antagonist strychnine, respectively. Mg2+ and DL-AP5, NMDA receptor blockers, CNQX, an antagonist of both the AMPA and kainate receptors, and 2-hydroxysaclofen, a GABAB receptor antagonist, do not affect quisqualamine's actions in vitro, suggesting that it does not directly affect the ionotropic glutamate receptors or the GABAB receptor in any way. Whether it binds to and acts upon any of the metabotropic glutamate receptors like its analogue quisqualic acid however is unclear.

In neuroscience, glutamate is the anion of glutamic acid in its role as a neurotransmitter. It is by a wide margin the most abundant excitatory neurotransmitter in the vertebrate nervous system. It is used by every major excitatory function in the vertebrate brain, accounting in total for well over 90% of the synaptic connections in the human brain. It also serves as the primary neurotransmitter for some localized brain regions, such as cerebellum granule cells.

Willardiine (correctly spelled with two successive i's) or (S)-1-(2-amino-2-carboxyethyl)pyrimidine-2,4-dione is a chemical compound that occurs naturally in the seeds of Mariosousa willardiana and Acacia sensu lato. The seedlings of these plants contain enzymes capable of complex chemical substitutions that result in the formation of free amino acids (See:#Synthesis). Willardiine is frequently studied for its function in higher level plants. Additionally, many derivates of willardiine are researched for their potential in pharmaceutical development. Willardiine was first discovered in 1959 by R. Gmelin, when he isolated several free, non-protein amino acids from Acacia willardiana (another name for Mariosousa willardiana) when he was studying how these families of plants synthesize uracilyalanines. A related compound, Isowillardiine, was concurrently isolated by a different group, and it was discovered that the two compounds had different structural and functional properties. Subsequent research on willardiine has focused on the functional significance of different substitutions at the nitrogen group and the development of analogs of willardiine with different pharmacokinetic properties. In general, Willardiine is the one of the first compounds studied in which slight changes to molecular structure result in compounds with significantly different pharmacokinetic properties.



