
Cardiomyopathy is a group of primary diseases of the heart muscle. Early on there may be few or no symptoms. As the disease worsens, shortness of breath, feeling tired, and swelling of the legs may occur, due to the onset of heart failure. An irregular heart beat and fainting may occur. Those affected are at an increased risk of sudden cardiac death.

Echocardiography, also known as cardiac ultrasound, is the use of ultrasound to examine the heart. It is a type of medical imaging, using standard ultrasound or Doppler ultrasound. The visual image formed using this technique is called an echocardiogram, a cardiac echo, or simply an echo.

Intermediate filaments (IFs) are cytoskeletal structural components found in the cells of vertebrates, and many invertebrates. Homologues of the IF protein have been noted in an invertebrate, the cephalochordate Branchiostoma.
An ejection fraction (EF) is the volumetric fraction of fluid ejected from a chamber with each contraction. It can refer to the cardiac atrium, ventricle, gall bladder, or leg veins, although if unspecified it usually refers to the left ventricle of the heart. EF is widely used as a measure of the pumping efficiency of the heart and is used to classify heart failure types. It is also used as an indicator of the severity of heart failure, although it has recognized limitations.

Arrhythmogenic cardiomyopathy (ACM), arrhythmogenic right ventricular dysplasia (ARVD), or arrhythmogenic right ventricular cardiomyopathy (ARVC), most commonly is an inherited heart disease.
Hypertrophic cardiomyopathy is a condition in which muscle tissues of the heart become thickened without an obvious cause. The parts of the heart most commonly affected are the interventricular septum and the ventricles. This results in the heart being less able to pump blood effectively and also may cause electrical conduction problems. Specifically, within the bundle branches that conduct impulses through the interventricular septum and into the Purkinje fibers, as these are responsible for the depolarization of contractile cells of both ventricles.

Dilated cardiomyopathy (DCM) is a condition in which the heart becomes enlarged and cannot pump blood effectively. Symptoms vary from none to feeling tired, leg swelling, and shortness of breath. It may also result in chest pain or fainting. Complications can include heart failure, heart valve disease, or an irregular heartbeat.
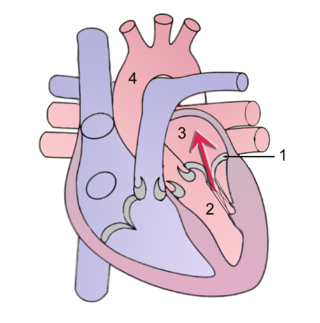
Mitral regurgitation(MR), also known as mitral insufficiency or mitral incompetence, is a form of valvular heart disease in which the mitral valve is insufficient and does not close properly when the heart pumps out blood. It is the abnormal leaking of blood backwards – regurgitation from the left ventricle, through the mitral valve, into the left atrium, when the left ventricle contracts. Mitral regurgitation is the most common form of valvular heart disease.

Peripartum cardiomyopathy (PPCM) is a form of dilated cardiomyopathy that is defined as a deterioration in cardiac function presenting typically between the last month of pregnancy and up to six months postpartum. As with other forms of dilated cardiomyopathy, PPCM involves systolic dysfunction of the heart with a decrease of the left ventricular ejection fraction (EF) with associated congestive heart failure and an increased risk of atrial and ventricular arrhythmias, thromboembolism (blockage of a blood vessel by a blood clot), and even sudden cardiac death. In essence, the heart muscle cannot contract forcefully enough to pump adequate amounts of blood for the needs of the body's vital organs.

Desmin is a protein that in humans is encoded by the DES gene. Desmin is a muscle-specific, type III intermediate filament that integrates the sarcolemma, Z disk, and nuclear membrane in sarcomeres and regulates sarcomere architecture.
Tachycardia-induced cardiomyopathy (TIC) is a disease where prolonged tachycardia or arrhythmia causes an impairment of the myocardium, which can result in heart failure. People with TIC may have symptoms associated with heart failure and/or symptoms related to the tachycardia or arrhythmia. Though atrial fibrillation is the most common cause of TIC, several tachycardias and arrhythmias have been associated with the disease.

Cardiac amyloidosis is a subcategory of amyloidosis where there is depositing of the protein amyloid in the cardiac muscle and surrounding tissues. Amyloid, a misfolded and insoluble protein, can become a deposit in the heart's atria, valves, or ventricles. These deposits can cause thickening of different sections of the heart, leading to decreased cardiac function. The overall decrease in cardiac function leads to a plethora of symptoms. This multisystem disease was often misdiagnosed, with a corrected analysis only during autopsy. Advancements of technologies have increased earlier accuracy of diagnosis. Cardiac amyloidosis has multiple sub-types including light chain, familial, and senile. One of the most studied types is light chain cardiac amyloidosis. Prognosis depends on the extent of the deposits in the body and the type of amyloidosis. New treatment methods are actively being researched in regards to the treatment of heart failure and specific cardiac amyloidosis problems.

Noncompaction cardiomyopathy (NCC) is a rare congenital disease of heart muscle that affects both children and adults. It results from abnormal prenatal development of heart muscle.

Alpha-crystallin B chain is a protein that in humans is encoded by the CRYAB gene. It is part of the small heat shock protein family and functions as molecular chaperone that primarily binds misfolded proteins to prevent protein aggregation, as well as inhibit apoptosis and contribute to intracellular architecture. Post-translational modifications decrease the ability to chaperone. Mutations in CRYAB cause different cardiomyopathies, skeletal myopathies mainly myofibrillar myopathy, and also cataracts. In addition, defects in this gene/protein have been associated with cancer and neurodegenerative diseases such as Alzheimer's disease and Parkinson's disease.

Diabetic cardiomyopathy is a disorder of the heart muscle in people with diabetes. It can lead to inability of the heart to circulate blood through the body effectively, a state known as heart failure(HF), with accumulation of fluid in the lungs or legs. Most heart failure in people with diabetes results from coronary artery disease, and diabetic cardiomyopathy is only said to exist if there is no coronary artery disease to explain the heart muscle disorder.
The E/A ratio is a marker of the function of the left ventricle of the heart. It represents the ratio of peak velocity blood flow from left ventricular relaxation in early diastole to peak velocity flow in late diastole caused by atrial contraction. It is calculated using Doppler echocardiography, an ultrasound-based cardiac imaging modality. Abnormalities in the E/A ratio suggest that the left ventricle, which pumps blood into the systemic circulation, cannot fill with blood properly in the period between contractions. This phenomenon is referred to as diastolic dysfunction and can eventually lead to the symptoms of heart failure.
Amyloid cardiomyopathy is a condition resulting in the death of part of the myocardium. It is associated with the systemic production and release of many amyloidogenic proteins, especially immunoglobulin light chain or transthyretin (TTR). It can be characterized by the extracellular deposition of amyloids, foldable proteins that stick together to build fibrils in the heart. The amyloid can be seen under polarized light in congo red stained biopsy.
Familial amyloid cardiomyopathy (FAC), or transthyretin amyloid cardiomyopathy (ATTR-CM) results from the aggregation and deposition of mutant and wild-type transthyretin (TTR) protein in the heart. TTR is usually circulated as a homo-tetramer—a protein made up of four identical subunits—however, in FAC populations, TTR dissociates from this typical form and misassembles into amyloid fibrils which are insoluble and resistant to degradation. Due to this resistance to degradation, when amyloid fibrils accumulate in the heart's walls, specifically the left ventricle, rigidity prevents the heart from properly relaxing and refilling with blood: this is called diastolic dysfunction which can ultimately lead to heart failure.

Heart failure with preserved ejection fraction (HFpEF) is a form of heart failure in which the ejection fraction – the percentage of the volume of blood ejected from the left ventricle with each heartbeat divided by the volume of blood when the left ventricle is maximally filled – is normal, defined as greater than 50%; this may be measured by echocardiography or cardiac catheterization. Approximately half of people with heart failure have preserved ejection fraction, while the other half have a reduction in ejection fraction, called heart failure with reduced ejection fraction (HFrEF).
The main pathophysiology of heart failure is a reduction in the efficiency of the heart muscle, through damage or overloading. As such, it can be caused by a wide number of conditions, including myocardial infarction, hypertension and cardiac amyloidosis. Over time these increases in workload will produce changes to the heart itself:
