Related Research Articles

Hemoglobinopathy is the medical term for a group of inherited blood disorders and diseases that primarily affect red blood cells. They are single-gene disorders and, in most cases, they are inherited as autosomal co-dominant traits.

Anemia or anaemia is a blood disorder in which the blood has a reduced ability to carry oxygen due to a lower than normal number of red blood cells, or a reduction in the amount of hemoglobin. The name is derived from Ancient Greek: ἀναιμία anaimia, meaning 'lack of blood', from ἀν- an-, 'not' and αἷμα haima, 'blood'. When anemia comes on slowly, the symptoms are often vague, such as tiredness, weakness, shortness of breath, headaches, and a reduced ability to exercise. When anemia is acute, symptoms may include confusion, feeling like one is going to pass out, loss of consciousness, and increased thirst. Anemia must be significant before a person becomes noticeably pale. Symptoms of anemia depend on how quickly hemoglobin decreases. Additional symptoms may occur depending on the underlying cause. Preoperative anemia can increase the risk of needing a blood transfusion following surgery. Anemia can be temporary or long term and can range from mild to severe.

Reticulocytes are immature red blood cells (RBCs). In the process of erythropoiesis, reticulocytes develop and mature in the bone marrow and then circulate for about a day in the blood stream before developing into mature red blood cells. Like mature red blood cells, in mammals, reticulocytes do not have a cell nucleus. They are called reticulocytes because of a reticular (mesh-like) network of ribosomal RNA that becomes visible under a microscope with certain stains such as new methylene blue and Romanowsky stain.

Thalassemias are inherited blood disorders that result in abnormal hemoglobin. Symptoms depend on the type of thalassemia and can vary from none to severe. Often there is mild to severe anemia as thalassemia can affect the production of red blood cells and also affect how long the red blood cells live. Symptoms of anemia include feeling tired and having pale skin. Other symptoms of thalassemia include bone problems, an enlarged spleen, yellowish skin, pulmonary hypertension, and dark urine. Slow growth may occur in children. Symptoms and presentations of thalassemia can change over time.

Hereditary spherocytosis (HS) is a congenital hemolytic disorder, wherein a genetic mutation coding for a structural membrane protein phenotype leads to a spherical shaping of erythrocytic cellular morphology. As erythrocytes are sphere-shaped (spherocytosis), rather than the normal biconcave disk-shaped, their morphology interferes with these cells' abilities to be flexible during circulation throughout the entirety of the body - arteries, arterioles, capillaries, venules, veins, and organs. This difference in shape also makes the red blood cells more prone to rupture under osmotic and/or mechanical stress. Cells with these dysfunctional proteins are degraded in the spleen, which leads to a shortage of erythrocytes resulting in hemolytic anemia.
The mean corpuscular volume, or mean cell volume (MCV), is a measure of the average volume of a red blood corpuscle. The measure is obtained by multiplying a volume of blood by the proportion of blood that is cellular, and dividing that product by the number of erythrocytes in that volume. The mean corpuscular volume is a part of a standard complete blood count.
Red blood cell distribution width (RDW), as well as various types thereof, is a measure of the range of variation of red blood cell (RBC) volume that is reported as part of a standard complete blood count. Red blood cells have an average volume of 80-100 femtoliters, but individual cell volumes vary even in healthy blood. Certain disorders, however, cause a significantly increased variation in cell size. Higher RDW values indicate greater variation in size. Normal reference range of RDW-CV in human red blood cells is 11.5–15.4%. If anemia is observed, RDW test results are often used together with mean corpuscular volume (MCV) results to determine the possible causes of the anemia. It is mainly used to differentiate an anemia of mixed causes from an anemia of a single cause.

Megaloblastic anemia is a type of macrocytic anemia. An anemia is a red blood cell defect that can lead to an undersupply of oxygen. Megaloblastic anemia results from inhibition of DNA synthesis during red blood cell production. When DNA synthesis is impaired, the cell cycle cannot progress from the G2 growth stage to the mitosis (M) stage. This leads to continuing cell growth without division, which presents as macrocytosis. Megaloblastic anemia has a rather slow onset, especially when compared to that of other anemias. The defect in red cell DNA synthesis is most often due to hypovitaminosis, specifically vitamin B12 deficiency or folate deficiency. Loss of micronutrients may also be a cause.
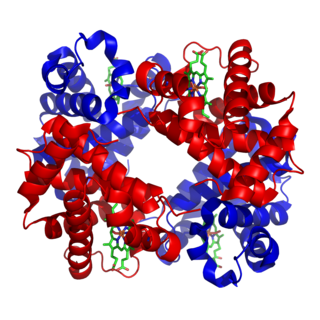
Hemoglobin A (HbA), also known as adult hemoglobin, hemoglobin A1 or α2β2, is the most common human hemoglobin tetramer, accounting for over 97% of the total red blood cell hemoglobin. Hemoglobin is an oxygen-binding protein, found in erythrocytes, which transports oxygen from the lungs to the tissues. Hemoglobin A is the most common adult form of hemoglobin and exists as a tetramer containing two alpha subunits and two beta subunits (α2β2). Hemoglobin A2 (HbA2) is a less common adult form of hemoglobin and is composed of two alpha and two delta-globin subunits. This hemoglobin makes up 1-3% of hemoglobin in adults.
Microcytic anaemia is any of several types of anemia characterized by smaller than normal red blood cells. The normal mean corpuscular volume is approximately 80–100 fL. When the MCV is <80 fL, the red cells are described as microcytic and when >100 fL, macrocytic. The MCV is the average red blood cell size.

Alpha-thalassemia is a form of thalassemia involving the genes HBA1 and HBA2. Thalassemias are a group of inherited blood conditions which result in the impaired production of hemoglobin, the molecule that carries oxygen in the blood. Normal hemoglobin consists of two alpha chains and two beta chains; in alpha-thalassemia, there is a quantitative decrease in the amount of alpha chains, resulting in fewer normal hemoglobin molecules. Furthermore, alpha-thalassemia leads to the production of unstable beta globin molecules which cause increased red blood cell destruction. The degree of impairment is based on which clinical phenotype is present.
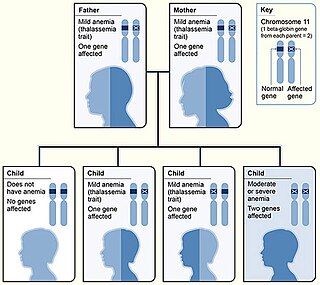
Beta thalassemias are a group of inherited blood disorders. They are forms of thalassemia caused by reduced or absent synthesis of the beta chains of hemoglobin that result in variable outcomes ranging from severe anemia to clinically asymptomatic individuals. Global annual incidence is estimated at one in 100,000. Beta thalassemias occur due to malfunctions in the hemoglobin subunit beta or HBB. The severity of the disease depends on the nature of the mutation.

Anisocytosis is a medical term meaning that a patient's red blood cells are of unequal size. This is commonly found in anemia and other blood conditions. False diagnostic flagging may be triggered on a complete blood count by an elevated WBC count, agglutinated RBCs, RBC fragments, giant platelets or platelet clumps. In addition, it is a characteristic feature of bovine blood.
Microcytosis or microcythemia is a condition in which red blood cells are unusually small as measured by their mean corpuscular volume.
Normocytic anemia is a type of anemia and is a common issue that occurs for men and women typically over 85 years old. Its prevalence increases with age, reaching 44 percent in men older than 85 years. The most common type of normocytic anemia is anemia of chronic disease.

Hemoglobin Lepore syndrome is typically an asymptomatic hemoglobinopathy, which is caused by an autosomal recessive genetic mutation. The Hb Lepore variant, consisting of two normal alpha globin chains (HBA) and two delta-beta globin fusion chains which occurs due to a "crossover" between the delta (HBD) and beta globin (HBB) gene loci during meiosis and was first identified in the Lepore family, an Italian-American family, in 1958. There are three varieties of Hb Lepore, Washington, Baltimore and Hollandia. All three varieties show similar electrophoretic and chromatographic properties and hematological findings bear close resemblance to those of the beta-thalassemia trait; a blood disorder that reduces the production of the iron-containing protein hemoglobin which carries oxygen to cells and which may cause anemia.
Treatment of the inherited blood disorder thalassemia depends upon the level of severity. For mild forms of the condition, advice and counseling are often all that are necessary. For more severe forms, treatment may consist in blood transfusion; chelation therapy to reverse iron overload, using drugs such as deferoxamine, deferiprone, or deferasirox; medication with the antioxidant indicaxanthin to prevent the breakdown of hemoglobin; or a bone marrow transplant using material from a compatible donor, or from the patient's mother. Removal of the spleen (splenectomy) could theoretically help to reduce the need for blood transfusions in people with thalassaemia major or intermedia but there is currently no reliable evidence from clinical trials about its effects. Population screening has had some success as a preventive measure.

A nucleated red blood cell (NRBC), also known by several other names, is a red blood cell that contains a cell nucleus. Almost all vertebrate organisms have hemoglobin-containing cells in their blood, and with the exception of mammals, all of these red blood cells are nucleated. In mammals, NRBCs occur in normal development as precursors to mature red blood cells in erythropoiesis, the process by which the body produces red blood cells.
Anemia is a condition in which blood has a lower-than-normal amount of red blood cells or hemoglobin. Anemia in pregnancy is a decrease in the total red blood cells (RBCs) or hemoglobin in the blood during pregnancy. Anemia is an extremely common condition in pregnancy world-wide, conferring a number of health risks to mother and child. While anemia in pregnancy may be pathologic, in normal pregnancies, the increase in RBC mass is smaller than the increase in plasma volume, leading to a mild decrease in hemoglobin concentration referred to as physiologic anemia. Maternal signs and symptoms are usually non-specific, but can include: fatigue, pallor, dyspnea, palpitations, and dizziness. There are numerous well-known maternal consequences of anemia including: maternal cardiovascular strain, reduced physical and mental performance, reduced peripartum blood reserves, increased risk for peripartum blood product transfusion, and increased risk for maternal mortality.

Transfusion-dependent anemia is a form of anemia characterized by the need for continuous blood transfusion. It is a condition that results from various diseases, and is associated with decreased survival rates. Regular transfusion is required to reduce the symptoms of anemia by increasing functional red blood cells and hemoglobin count. Symptoms may vary based on the severity of the condition and the most common symptom is fatigue. Various diseases can lead to transfusion-dependent anemia, most notably myelodysplastic syndromes (MDS) and thalassemia. Due to the number of diseases that can cause transfusion-dependent anemia, diagnosing it is more complicated. Transfusion dependence occurs when an average of more than 2 units of blood transfused every 28 days is required over a period of at least 3 months. Myelodysplastic syndromes is often only diagnosed when patients become anemic, and transfusion-dependent thalassemia is diagnosed based on gene mutations. Screening for heterozygosity in the thalassemia gene is an option for early detection.
References
- ↑ Mentzer WC (April 1973). "Differentiation of iron deficiency from thalassaemia trait". Lancet. 1 (7808): 882. doi:10.1016/S0140-6736(73)91446-3. PMID 4123424.
- ↑ Ntaios G, Chatzinikolaou A, Saouli Z, et al. (July 2007). "Discrimination indices as screening tests for beta-thalassemic trait". Ann. Hematol. 86 (7): 487–91. doi:10.1007/s00277-007-0302-x. PMID 17476506. S2CID 10499303.
- 1 2 "Beta Thalassemia Differential Diagnoses". emedicine.medscape.com. Retrieved 2023-06-28.
- ↑ Joseph Mazza (15 January 2002). Manual of clinical hematology. Lippincott Williams & Wilkins. pp. 152–. ISBN 978-0-7817-2980-2 . Retrieved 4 June 2010.
- ↑ "Mentzer Index". reference.medscape.com. Retrieved 2023-06-28.
| | This medical diagnostic article is a stub. You can help Wikipedia by expanding it. |