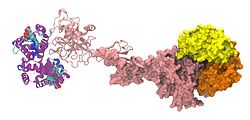
A model of α,β-hemoglobin/haptoglobin hexamer complex. There are 2 α,β-hemoglobin dimers depicted: one space filling model (yellow/orange), and one ribbon model (purple/blue). Each is bound by a haptoglobin molecule (both haptoglobin molecules are shown in pink, with one as a space filling model and one as a ribbon model).
In clinical settings, the haptoglobin assay is used to screen for and monitor intravascular hemolytic anemia. In intravascular hemolysis, free hemoglobin will be released into circulation and hence haptoglobin will bind the hemoglobin. This causes a decline in haptoglobin levels.
The protein was discovered as a "plasma substance" in 1938 by French biochemists Max-Fernand Jayle and Michel Polonovski.[9][10]
Function
Hemoglobin that has been released into the blood plasma by damaged red blood cells has harmful effects. The HP gene encodes a preproprotein that is processed to yield both alpha and beta chains, which subsequently combines as a tetramer to produce haptoglobin. Haptoglobin functions to bind the free plasma hemoglobin, which allows degradative enzymes to gain access to the hemoglobin while at the same time preventing loss of iron through the kidneys and protecting the kidneys from damage by hemoglobin.[11]
The cellular receptor target of Hp is the monocyte/macrophage scavenger receptor, CD163.[7] Following Hb-Hp binding to CD163, cellular internalization of the complex leads to globin and heme metabolism, which is followed by adaptive changes in antioxidant and iron metabolism pathways and macrophage phenotype polarization.[7]
Hp has hemoglobin-independent immunomodulatory functions. It dampens lipopolysaccharide-induced cytokine expression.[12] Lipopolysaccharides directly bind to Hp, which, due to the high abundance of Hp in serum, results in their buffering and shielding from toll-like receptor 4. Functionally, this results in delayed activation of the NF-κB pathway.[13]
Difference from hemopexin
When hemoglobin is released from RBCs within the physiologic range of haptoglobin, the potential deleterious effects of hemoglobin are prevented. During hyper-hemolytic conditions or with chronic hemolysis, haptoglobin is depleted and hemoglobin readily distributes to tissues where it might be exposed to oxidative conditions. In such conditions, heme can be released from ferric (Fe3+-bound) hemoglobin. The free heme can then accelerate tissue damage by promoting peroxidative reactions and activation of inflammatory cascades. Hemopexin (Hx) is another plasma glycoprotein that is able to bind heme with high affinity. It sequesters heme in an inert non-toxic form and transports it to the liver for catabolism and excretion.[7]
Haptoglobin had been shown to be expressed in adipose tissue of cattle as well.[15]
Structure
Haptoglobin, in its simplest form, consists of two alpha and two beta chains, connected by disulfide bridges. The chains originate from a common precursor protein, which is proteolytically cleaved during protein synthesis.
Hp exists in two allelic forms in the human population, so-called Hp1 and Hp2, the latter one having arisen due to the partial duplication of Hp1 gene. Three genotypes of Hp, therefore, are found in humans: Hp1-1, Hp2-1, and Hp2-2. Hp of different genotypes have been shown to bind hemoglobin with different affinities, with Hp2-2 being the weakest binder. Allele 2 encodes for two multimerization domains. This results in oligomer formation in carriers of allele 2.[5] The frequency of the short allele varies between 7% and 70% depending on racial origin.[16] It is unclear which evolutionary advantage is conferred by the longer allele; strikingly, a similar partial duplication independently arose much earlier in a precursor of ruminants. Ruminants exclusively express oligomeric haptoglobin.[17]
Since the reticuloendothelial system will remove the haptoglobin-hemoglobin complex from the body,[8] haptoglobin levels will be decreased in case of intravascular hemolysis or severeextravascular hemolysis. In the process of binding to free hemoglobin, haptoglobin sequesters the iron within hemoglobin, preventing iron-utilizing bacteria from benefiting from hemolysis. It is theorized that, because of this, haptoglobin has evolved into an acute-phase protein. HP has a protective influence on the hemolytic kidney.[25][26]
The different haptoglobin phenotypes differ in their antioxidant, scavenging,[27] and immunomodulatory properties. This aspect of haptoglobin may gain importance in immune suppressed conditions (such as liver cirrhosis) and the various phenotypes may result in different susceptibility levels towards bacterial infections.[28]
Some studies associate certain haptoglobin phenotypes with the risk of developing hypertension in diabetes[29] and schizophrenia.[30]
Test protocol
Measuring the level of haptoglobin in a patient's blood is ordered whenever a patient exhibits symptoms of anemia, such as pallor, fatigue, or shortness of breath, along with physical signs of hemolysis, such as jaundice or dark-colored urine. The test is also commonly ordered as a hemolytic anemia battery, which also includes a reticulocyte count and a peripheral blood smear. It can also be ordered along with a direct antiglobulin test when a patient is suspected of having a transfusion reaction or symptoms of autoimmune hemolytic anemia. Also, it may be ordered in conjunction with a bilirubin.
Interpretation
A decrease in haptoglobin can support a diagnosis of hemolysis, especially when correlated with a decreased hemoglobin, and hematocrit, and also an increased reticulocyte count. Low haptoglobin levels occur regardless of the site and mechanism of haemolysis (intravascular and splenic/"extravascular") [31]
If the reticulocyte count is increased, but the haptoglobin level is normal, this argues against haemolysis, and suggests a bone marrow response to blood loss. If there are symptoms of anemia but both the reticulocyte count and the haptoglobin level are normal, the anemia is most likely not due to hemolysis. Haptoglobin levels that are decreased but do not accompany signs of anemia may indicate advanced liver damage, as the liver is the major site of production of haptoglobin.
As haptoglobin is an acute-phase protein, any inflammatory process (infection, injury, allergy, etc.) may increase the levels of plasma haptoglobin, but patients with haemolysis usually have low haptoglobin regardless of the presence of inflammation [31]
↑ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
↑ "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
1 2 Dobryszycka W (September 1997). "Biological functions of haptoglobin--new pieces to an old puzzle". European Journal of Clinical Chemistry and Clinical Biochemistry. 35 (9): 647–654. PMID9352226.
↑ Wassell J (2000). "Haptoglobin: function and polymorphism". Clinical Laboratory. 46 (11–12): 547–552. PMID11109501.
↑ Saremi B, Al-Dawood A, Winand S, Müller U, Pappritz J, von Soosten D, etal. (May 2012). "Bovine haptoglobin as an adipokine: serum concentrations and tissue expression in dairy cows receiving a conjugated linoleic acids supplement throughout lactation". Veterinary Immunology and Immunopathology. 146 (3–4): 201–211. doi:10.1016/j.vetimm.2012.03.011. PMID22498004.
↑ Sadrzadeh SM, Bozorgmehr J (June 2004). "Haptoglobin phenotypes in health and disorders". American Journal of Clinical Pathology. 121 (Suppl): S97-104. doi:10.1309/8GLX5798Y5XHQ0VW. PMID15298155.
↑ Papp M, Lakatos PL, Palatka K, Foldi I, Udvardy M, Harsfalvi J, etal. (May 2007). "Haptoglobin polymorphisms are associated with Crohn's disease, disease behavior, and extraintestinal manifestations in Hungarian patients". Digestive Diseases and Sciences. 52 (5): 1279–1284. doi:10.1007/s10620-006-9615-1. PMID17357835. S2CID35999438.
↑ Costa-Mallen P, Checkoway H, Zabeti A, Edenfield MJ, Swanson PD, Longstreth WT, etal. (March 2008). "The functional polymorphism of the hemoglobin-binding protein haptoglobin influences susceptibility to idiopathic Parkinson's disease". American Journal of Medical Genetics. Part B, Neuropsychiatric Genetics. 147B (2): 216–222. doi:10.1002/ajmg.b.30593. PMID17918239. S2CID21568460.
↑ Pintera J (1968). "The protective influence of haptoglobin on hemoglobinuric kidney. I. Bioch- emical and macroscopic observations". Folia Haematologica. 90 (1): 82–91. PMID4176393.
↑ Miederer SE, Hotz J (December 1969). "[Pathogenesis of kidney hemolysis]". Bruns' Beiträge für Klinische Chirurgie (in German). 217 (7): 661–665. PMID5404273.
↑ Vitalis Z, Altorjay I, Tornai I, Palatka K, Kacska S, Palyu E, etal. (April 2011). "Phenotypic polymorphism of haptoglobin: a novel risk factor for the development of infection in liver cirrhosis". Human Immunology. 72 (4): 348–354. doi:10.1016/j.humimm.2011.01.008. PMID21262313.
1 2 Körmöczi GF, Säemann MD, Buchta C, Peck-Radosavljevic M, Mayr WR, Schwartz DW, etal. (March 2006). "Influence of clinical factors on the haemolysis marker haptoglobin". European Journal of Clinical Investigation. 36 (3): 202–209. doi:10.1111/j.1365-2362.2006.01617.x. PMID16506966. S2CID39884908.
Further reading
Graversen JH, Madsen M, Moestrup SK (April 2002). "CD163: a signal receptor scavenging haptoglobin-hemoglobin complexes from plasma". The International Journal of Biochemistry & Cell Biology. 34 (4): 309–314. doi:10.1016/S1357-2725(01)00144-3. PMID11854028.
Erickson LM, Kim HS, Maeda N (December 1992). "Junctions between genes in the haptoglobin gene cluster of primates". Genomics. 14 (4): 948–958. doi:10.1016/S0888-7543(05)80116-8. PMID1478675.
Simmers RN, Stupans I, Sutherland GR (1986). "Localization of the human haptoglobin genes distal to the fragile site at 16q22 using in situ hybridization". Cytogenetics and Cell Genetics. 41 (1): 38–41. doi:10.1159/000132193. PMID3455911.
van der Straten A, Falque JC, Loriau R, Bollen A, Cabezón T (April 1986). "Expression of cloned human haptoglobin and alpha 1-antitrypsin complementary DNAs in Saccharomyces cerevisiae". DNA. 5 (2): 129–136. doi:10.1089/dna.1986.5.129. PMID3519135.
Malchy B, Dixon GH (March 1973). "Studies on the interchain disulfides of human haptoglobins". Canadian Journal of Biochemistry. 51 (3): 249–264. doi:10.1139/o73-032. PMID4573324.
Tabak S, Lev A, Valansi C, Aker O, Shalitin C (November 1996). "Transcriptionally active haptoglobin-related (Hpr) gene in hepatoma G2 and leukemia molt-4 cells". DNA and Cell Biology. 15 (11): 1001–1007. doi:10.1089/dna.1996.15.1001. PMID8945641.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.