Myelin protein zero (MPZ), also Myelin protein P0, is a single membrane glycoprotein[5] which in humans is encoded by the MPZ gene. P0 is a major structural component of the myelin sheath in the peripheral nervous system (PNS).[6] Myelin protein zero is expressed by Schwann cells and accounts for over 50% of all proteins in the peripheral nervous system, making it the most common protein expressed in the PNS.[6] Mutations in myelin protein zero can cause myelin deficiency and are associated with neuropathies like Charcot–Marie–Tooth disease and Dejerine–Sottas disease.[7]
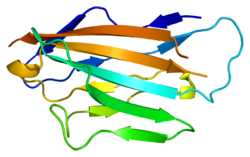
Structure of myelin protein zero's extracellular domain with labelled beta strands. Strands D, E, B, and A make up one beta sheet, Strands A', G, F, C, C', C'' make up the other beta sheet.
In humans, the gene that encodes myelin protein zero is located on chromosome 1 near the Duffy locus or the Duffy antigen/chemokine receptor. The gene is about 7,000 bases long and is divided into 6 exons. In total, myelin protein zero is 219 amino acids long[6] and has many basic amino acid residues.[8]
Myelin protein zero consists of an extracellular N-terminal domain (amino acids 1–124), a single transmembrane region (125–150), and a smaller positively charged intracellular region (151–219).[6][9][10] Its cytoplasmic domain is highly positively charged but presumably does not fold into a globular structure.[11] The extracellular domain is structurally similar to the immunoglobulin domain[8] and therefore the protein is considered as belonging to immunoglobulin superfamily.[12]
Besides existing as a monomer, myelin protein zero is also known to form dimers and tetramers with other myelin protein zero molecules in vertebrates.[13]
Function
The myelin sheath is a multi-layered membrane, unique to the nervous system, that functions as an insulator to greatly increase the velocity of axonal impulse conduction. Myelin protein zero, absent in the central nervous system,[14] is a major component of the myelin sheath in peripheralnerves. Mutations that disrupt the function of myelin protein zero can lead to less expression of myelin and degeneration of myelin sheath in the peripheral nervous system.[15] Currently, myelin protein zero expression is postulated to be produced by signals from the axon. However, more details about the regulation of myelin protein zero are unknown.[6]
It is postulated that myelin protein zero is a structural element in the formation and stabilization of peripheral nerve myelin.[9] Myelin protein zero is also hypothesized to serve as a cell adhesion molecule, holding multiple layers of myelin together.[10] When a myelinating cell wraps its membrane around an axon multiple times, generating multiple layers of myelin, myelin protein zero helps keep these sheets compact by serving as a "glue" that keeps the layers of myelin together.[11] It does so by holding its characteristic coil structure together by the electrostatic interactions[8] of its positively charged intracellular domain with acidiclipids in the cytoplasmic face of the opposite bilayer.[14] and by interaction between hydrophobic globular 'heads' of adjacent extracellular domains.[14]
Myelin protein zero's function is similar to the function of other proteins with immunoglobin domains like polyimmunoglobin and T4 protein. These proteins function as binding and adhesion molecules and participate in homotypic interactions, or interactions that involve two similar proteins.[9] Myelin protein zero holds together the myelin sheath by participating in homotypic interactions with other myelin protein zero proteins. Myelin protein zero's extracellular domain binds to the myelin sphingolipid membrane and holds together myelin layers using homotypic interactions with other myelin protein zero extracellular domains,[7] and with extracellular tryptophan residues interacting with the membrane.[8]
Myelin protein zero has also been demonstrated to interact with other proteins like peripheral myelin protein 22.[16] However, at this point the purpose of these interactions has not yet been determined.[16]
Associations with neuropathy
Mutations in myelin protein zero are known to cause myelin degeneration and neuropathy.[7] Mutations that reduce myelin protein zero's adhesion function or its ability to participate in homeotypic interactions with other myelin protein zero proteins are thought to cause neuropathy.[17] Mutations to myelin protein zero can lead to issues with the development of myelin early on in life or myelin degeneration on the axon later on in life.[12] Some mutations can cause neuropathy in infancy like Derjerine-Sottas disease while other mutations can cause neuropathy within the first two decades of life like Charcot-Marie-Tooth disease.[7] Adding a charged amino acid or changing a cysteine residue in the extracellular membrane can lead to neuropathy onset early on. Truncating the cytoplasmic domain or changing the tertiary structure of myelin protein zero can also result in neuropathy[7] because the cytoplasmic domain has been demonstrated to be necessary for homotypic interactions.[12]
1 2 Han H, Myllykoski M, Ruskamo S, Wang C, Kursula P (January 2013). "Myelin-specific proteins: a structurally diverse group of membrane-interacting molecules". BioFactors. 39 (3): 233–41. doi:10.1002/biof.1076. PMID23780694. S2CID21111930.
↑ Thompson AJ, Cronin MS, Kirschner DA (March 2002). "Myelin protein zero exists as dimers and tetramers in native membranes of Xenopus laevis peripheral nerve". Journal of Neuroscience Research. 67 (6): 766–71. doi:10.1002/jnr.10167. PMID11891790. S2CID36556147.
Patel PI, Lupski JR (April 1994). "Charcot-Marie-Tooth disease: a new paradigm for the mechanism of inherited disease". Trends in Genetics. 10 (4): 128–33. doi:10.1016/0168-9525(94)90214-3. PMID7518101.
Watanabe M, Yamamoto N, Ohkoshi N, Nagata H, Kohno Y, Hayashi A, etal. (September 2002). "Corticosteroid- responsive asymmetric neuropathy with a myelin protein zero gene mutation". Neurology. 59 (5): 767–9. doi:10.1212/wnl.59.5.767. PMID12221176.
Hayasaka K, Nanao K, Tahara M, Sato W, Takada G, Miura M, etal. (October 1991). "Isolation and sequence determination of cDNA encoding the major structural protein of human peripheral myelin". Biochemical and Biophysical Research Communications. 180 (2): 515–8. doi:10.1016/S0006-291X(05)81094-0. PMID1719967.
Ouvrier RA, McLeod JG, Conchin TE (February 1987). "The hypertrophic forms of hereditary motor and sensory neuropathy. A study of hypertrophic Charcot-Marie-Tooth disease (HMSN type I) and Dejerine-Sottas disease (HMSN type III) in childhood". Brain. 110 ( Pt 1) (1): 121–48. doi:10.1093/brain/110.1.121. PMID3467805.
Hayasaka K, Himoro M, Wang Y, Takata M, Minoshima S, Shimizu N, etal. (September 1993). "Structure and chromosomal localization of the gene encoding the human myelin protein zero (MPZ)". Genomics. 17 (3): 755–8. doi:10.1006/geno.1993.1400. PMID7503936.
Himoro M, Yoshikawa H, Matsui T, Mitsui Y, Takahashi M, Kaido M, etal. (September 1993). "New mutation of the myelin P0 gene in a pedigree of Charcot-Marie-Tooth neuropathy 1". Biochemistry and Molecular Biology International. 31 (1): 169–73. PMID7505151.
Hayasaka K, Himoro M, Sawaishi Y, Nanao K, Takahashi T, Takada G, etal. (November 1993). "De novo mutation of the myelin P0 gene in Dejerine-Sottas disease (hereditary motor and sensory neuropathy type III)". Nature Genetics. 5 (3): 266–8. doi:10.1038/ng1193-266. PMID7506095. S2CID2512684.
Pham-Dinh D, Fourbil Y, Blanquet F, Mattéi MG, Roeckel N, Latour P, etal. (December 1993). "The major peripheral myelin protein zero gene: structure and localization in the cluster of Fc gamma receptor genes on human chromosome 1q21.3-q23". Human Molecular Genetics. 2 (12): 2051–4. doi:10.1093/hmg/2.12.2051. PMID7509228.
Thomas FP, Lebo RV, Rosoklija G, Ding XS, Lovelace RE, Latov N, etal. (1994). "Tomaculous neuropathy in chromosome 1 Charcot-Marie-Tooth syndrome". Acta Neuropathologica. 87 (1): 91–7. doi:10.1007/BF00386259. PMID7511317. S2CID19827120.
Nelis E, Timmerman V, De Jonghe P, Vandenberghe A, Pham-Dinh D, Dautigny A, etal. (December 1994). "Rapid screening of myelin genes in CMT1 patients by SSCP analysis: identification of new mutations and polymorphisms in the P0 gene". Human Genetics. 94 (6): 653–7. doi:10.1007/bf00206959. PMID7527371. S2CID5750189.
Hilmi S, Fournier M, Valeins H, Gandar JC, Bonnet J (February 1995). "Myelin P0 glycoprotein: identification of the site phosphorylated in vitro and in vivo by endogenous protein kinases". Journal of Neurochemistry. 64 (2): 902–7. doi:10.1046/j.1471-4159.1995.64020902.x. PMID7530295. S2CID32511382.
Rautenstrauss B, Nelis E, Grehl H, Pfeiffer RA, Van Broeckhoven C (September 1994). "Identification of a de novo insertional mutation in P0 in a patient with a Déjérine-Sottas syndrome (DSS) phenotype". Human Molecular Genetics. 3 (9): 1701–2. doi:10.1093/hmg/3.9.1701. PMID7530550.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.