Autocrine signaling is a form of cell signaling in which a cell secretes a hormone or chemical messenger that binds to autocrine receptors on that same cell, leading to changes in the cell. This can be contrasted with paracrine signaling, intracrine signaling, or classical endocrine signaling.

Cadherins (named for "calcium-dependent adhesion") are cell adhesion molecules important in forming adherens junctions that let cells adhere to each other. Cadherins are a class of type-1 transmembrane proteins, and they depend on calcium (Ca2+) ions to function, hence their name. Cell-cell adhesion is mediated by extracellular cadherin domains, whereas the intracellular cytoplasmic tail associates with numerous adaptors and signaling proteins, collectively referred to as the cadherin adhesome.
Catenins are a family of proteins found in complexes with cadherin cell adhesion molecules of animal cells. The first two catenins that were identified became known as α-catenin and β-catenin. α-Catenin can bind to β-catenin and can also bind filamentous actin (F-actin). β-Catenin binds directly to the cytoplasmic tail of classical cadherins. Additional catenins such as γ-catenin and δ-catenin have been identified. The name "catenin" was originally selected because it was suspected that catenins might link cadherins to the cytoskeleton.

The selectins are a family of cell adhesion molecules. All selectins are single-chain transmembrane glycoproteins that share similar properties to C-type lectins due to a related amino terminus and calcium-dependent binding. Selectins bind to sugar moieties and so are considered to be a type of lectin, cell adhesion proteins that bind sugar polymers.
The epithelial–mesenchymal transition (EMT) is a process by which epithelial cells lose their cell polarity and cell–cell adhesion, and gain migratory and invasive properties to become mesenchymal stem cells; these are multipotent stromal cells that can differentiate into a variety of cell types. EMT is essential for numerous developmental processes including mesoderm formation and neural tube formation. EMT has also been shown to occur in wound healing, in organ fibrosis and in the initiation of metastasis in cancer progression.
Perlecan (PLC) also known as basement membrane-specific heparan sulfate proteoglycan core protein (HSPG) or heparan sulfate proteoglycan 2 (HSPG2), is a protein that in humans is encoded by the HSPG2 gene. The HSPG2 gene codes for a 4,391 amino acid protein with a molecular weight of 468,829. It is one of the largest known proteins. The name perlecan comes from its appearance as a "string of pearls" in rotary shadowed images.

E-selectin, also known as CD62 antigen-like family member E (CD62E), endothelial-leukocyte adhesion molecule 1 (ELAM-1), or leukocyte-endothelial cell adhesion molecule 2 (LECAM2), is a selectin cell adhesion molecule expressed only on endothelial cells activated by cytokines. Like other selectins, it plays an important part in inflammation. In humans, E-selectin is encoded by the SELE gene.

CD146 also known as the melanoma cell adhesion molecule (MCAM) or cell surface glycoprotein MUC18, is a 113kDa cell adhesion molecule currently used as a marker for endothelial cell lineage. In humans, the CD146 protein is encoded by the MCAM gene.

Syndecans are single transmembrane domain proteins that are thought to act as coreceptors, especially for G protein-coupled receptors. More specifically, these core proteins carry three to five heparan sulfate and chondroitin sulfate chains, i.e. they are proteoglycans, which allow for interaction with a large variety of ligands including fibroblast growth factors, vascular endothelial growth factor, transforming growth factor-beta, fibronectin and antithrombin-1. Interactions between fibronectin and some syndecans can be modulated by the extracellular matrix protein tenascin C.

72 kDa type IV collagenase also known as matrix metalloproteinase-2 (MMP-2) and gelatinase A is an enzyme that in humans is encoded by the MMP2 gene. The MMP2 gene is located on chromosome 16 at position 12.2.

Cluster of differentiation 97 is a protein also known as BL-Ac[F2] encoded by the ADGRE5 gene. CD97 is a member of the adhesion G protein-coupled receptor (GPCR) family. Adhesion GPCRs are characterized by an extended extracellular region often possessing N-terminal protein modules that is linked to a TM7 region via a domain known as the GPCR-Autoproteolysis INducing (GAIN) domain.

Cadherin-3, also known as P-Cadherin, is a protein that in humans is encoded by the CDH3 gene.

Epithelial cell adhesion molecule (EpCAM), also known as CD326 among other names, is a transmembrane glycoprotein mediating Ca2+-independent homotypic cell–cell adhesion in epithelia. EpCAM is also involved in cell signaling, migration, proliferation, and differentiation. Additionally, EpCAM has oncogenic potential via its capacity to upregulate c-myc, e-fabp, and cyclins A & E. Since EpCAM is expressed exclusively in epithelia and epithelial-derived neoplasms, EpCAM can be used as diagnostic marker for various cancers. It appears to play a role in tumorigenesis and metastasis of carcinomas, so it can also act as a potential prognostic marker and as a potential target for immunotherapeutic strategies.
Zinc finger protein SNAI1 is a protein that in humans is encoded by the SNAI1 gene. Snail is a family of transcription factors that promote the repression of the adhesion molecule E-cadherin to regulate epithelial to mesenchymal transition (EMT) during embryonic development.
Receptor-type tyrosine-protein phosphatase beta or VE-PTP is an enzyme specifically expressed in endothelial cells that in humans is encoded by the PTPRB gene.

Metastasis-associated protein MTA3 is a protein that in humans is encoded by the MTA3 gene. MTA3 protein localizes in the nucleus as well as in other cellular compartments MTA3 is a component of the nucleosome remodeling and deacetylate (NuRD) complex and participates in gene expression. The expression pattern of MTA3 is opposite to that of MTA1 and MTA2 during mammary gland tumorigenesis. However, MTA3 is also overexpressed in a variety of human cancers.

Sulfatase 1, also known as SULF1, is an enzyme which in humans is encoded by the SULF1 gene.

Cadherin-1 or Epithelial cadherin(E-cadherin), is a protein that in humans is encoded by the CDH1 gene. Mutations are correlated with gastric, breast, colorectal, thyroid, and ovarian cancers. CDH1 has also been designated as CD324. It is a tumor suppressor gene.
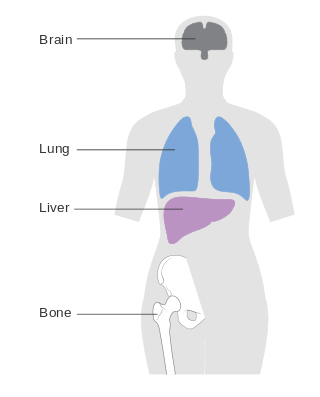
Metastatic breast cancer, also referred to as metastases, advanced breast cancer, secondary tumors, secondaries or stage IV breast cancer, is a stage of breast cancer where the breast cancer cells have spread to distant sites beyond the axillary lymph nodes. There is no cure for metastatic breast cancer; there is no stage after IV.

Vasculogenic mimicry (VM) is a strategy used by tumors to ensure sufficient blood supply is brought to its cells through establishing new tumor vascularization. This process is similar to tumor angiogenesis; on the other hand vascular mimicry is unique in that this process occurs independent of endothelial cells. Vasculature is instead developed de novo by cancer cells, which under stress conditions such as hypoxia, express similar properties to stem cells, capable of differentiating to mimic the function of endothelial cells and form vasculature-like structures. The ability of tumors to develop and harness nearby vasculature is considered one of the hallmarks of cancer disease development and is thought to be closely linked to tumor invasion and metastasis. Vascular mimicry has been observed predominantly in aggressive and metastatic cancers and has been associated with negative tumor characteristics such as increased metastasis, increased tissue invasion, and overall poor outcomes for patient survival. Vascular mimicry poses a serious problem for current therapeutic strategies due to its ability to function in the presence of Anti-angiogenic therapeutic agents. In fact, such therapeutics have been found to actually drive VM formation in tumors, causing more aggressive and difficult to treat tumors to develop.





