While the primary molecular function of MOG is not yet known, its likely role with the myelin sheath is either in sheath "completion and/or maintenance".[7] More specifically, MOG is speculated to be "necessary" as an "adhesion molecule" on the myelin sheath of the CNS to provide the structural integrity of the myelin sheath.[8][full citation needed]"
MOG's cDNA coding region in humans have been shown to be "highly homologous"[9] to rats, mice, and bovine, and hence highly conserved. This suggests "an important biological role for this protein".[7]
Physiology
The gene for MOG, found on chromosome 6 p21.3-p22,[10] was first sequenced in 1995. It is a transmembrane proteinexpressed on the surface of oligodendrocyte cell and on the outermost surface of myelin sheaths. "MOG is a quantitatively minor type I transmembrane protein,[11] and is found exclusively in the CNS. "A single Ig-domain is exposed to the extracellular space"[11] and consequently allows autoantibodies easy access. and therefore is easily accessible to autoantibodies too.[7][11] The MOG "primary nuclear transcript … is 15,561 nucleotides in length"[7] and, for humans, it has eight exons which are "separated by seven introns".[7] The introns "contain numerous reptitive[sic] DNA[7]" sequences, among which are "14 Alu sequences within 3 introns",[7] and have a range varying from 242 to 6484 bp.
Structure
Alternatively spliced human mRNA of the MOG gene form at least nine isoforms.[12]
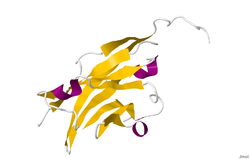
The crystal structure of myelin oligodendrocyte glycoprotein was determined by x-ray diffraction at a resolution of 1.45 Angstroms, using protein from the Norway rat. This protein is 139 residues long, and is a member of the immunoglobulin superfamily.[13] The dssp secondary structure of the protein is 6% helical and 43% beta sheet: there are three short helical segments and ten beta strands.[14] The beta strands are within two antiparallel beta sheets that form an immunoglobulin-like beta-sandwich fold.[15] Several features of the protein structure suggest MOG has a role as an "adhesin in the completion and/or compaction of the myelin sheath." There is a "significant strip" of electronegative charge beginning near the N-terminus and running about half the length of the molecule. Also, MOG was shown to dimerize in solution, and the shape complementarity index is high at the dimer interface, suggesting a "biologically relevant MOG dimer."[16]
Synthesis
Developmentally, MOG is formed "very late on oligodendrocytes and the myelin sheath".[8][full citation needed]
MOG has received much of its laboratory attention in studies dealing with MS. Several studies have shown a role for antibodies against MOG in the pathogenesis of MS,[8][full citation needed][18] though most of them were written before the discovery of NMO-IgG and the NMO spectrum of diseases.
Anti-MOG status is different depending whether it is measured by ELISA or by microarray (CBA). The proper way to identify it is by microarray, reacting patient serum with living cells, and detecting the binding IgG via a fluorescent-labeled secondary antibody.[19]
In animal models
Animal models of MS, namely Experimental Autoimmune Encephalomyelitis (EAE) models, have shown that "MOG-specific EAE models (of different animal strains) display/mirror human multiple sclerosis",[8][full citation needed] but basically explains the part involved in the optic neuritis.[20] These models with anti-MOG antibodies have been investigated extensively and are considered the only antibodies with demyelinating capacity[8][full citation needed] but again, EAE pathology is closer to NMO and ADEM than to the confluent demyelination observed in MS.
Anti-MOG antibodies have been shown to behave similarly to AQP4 antibodies in animal models,[20] and are considered a biomarker against the MS diagnosis[21][22]
↑ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
↑ "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
↑ Pham-Dinh D, Della Gaspera B, Kerlero de Rosbo N, Dautigny A (September 1995). "Structure of the human myelin/oligodendrocyte glycoprotein gene and multiple alternative spliced isoforms". Genomics. 29 (2): 345–52. doi:10.1006/geno.1995.9995. PMID8666381.
↑ Pham-Dinh D, Jones EP, Pitiot G, Della Gaspera B, Daubas P, Mallet J, Le Paslier D, Fischer Lindahl K, Dautigny A (1995). "Physical mapping of the human and mouse MOG gene at the distal end of the MHC class Ib region". Immunogenetics. 42 (5): 386–91. doi:10.1007/bf00179400. PMID7590972. S2CID8310478.
1 2 3 4 5 6 7 8 Roth MP, Malfroy L, Offer C, Sevin J, Enault G, Borot N, Pontarotti P, Coppin H (July 1995). "The human myelin oligodendrocyte glycoprotein (MOG) gene: complete nucleotide sequence and structural characterization". Genomics. 28 (2): 241–50. doi:10.1006/geno.1995.1137. PMID8530032.
1 2 3 4 5 6 Berger, T., Innsbruck Medical University Dept. of Neurology interviewed by S. Gillooly, Nov. 24, 2008.
↑ Pham-Dinh D, Allinquant B, Ruberg M, Della Gaspera B, Nussbaum JL, Dautigny A (December 1994). "Characterization and expression of the cDNA coding for the human myelin/oligodendrocyte glycoprotein". Journal of Neurochemistry. 63 (6): 2353–6. doi:10.1046/j.1471-4159.1994.63062353.x. PMID7964757. S2CID2788720.
↑ Kabsch W, Sander C (December 1983). "Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features". Biopolymers. 22 (12): 2577–637. doi:10.1002/bip.360221211. PMID6667333. S2CID29185760.
↑ Murzin AG, Brenner SE, Hubbard T, Chothia C (April 1995). "SCOP: a structural classification of proteins database for the investigation of sequences and structures". Journal of Molecular Biology. 247 (4): 536–40. doi:10.1016/S0022-2836(05)80134-2. PMID7723011.
↑ Berger T, Reindl M (August 2015). "Antibody biomarkers in CNS demyelinating diseases - a long and winding road". European Journal of Neurology. 22 (8): 1162–8. doi:10.1111/ene.12759. PMID26010364. S2CID39301229.
1 2 3 Reindl M, Di Pauli F, Rostásy K, Berger T (August 2013). "The spectrum of MOG autoantibody-associated demyelinating diseases". Nature Reviews. Neurology. 9 (8): 455–61. doi:10.1038/nrneurol.2013.118. PMID23797245. S2CID7219279.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.