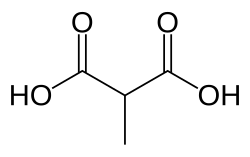
 | |
| Names | |
|---|---|
| Preferred IUPAC name Methylpropanedioic acid | |
| Other names Methylmalonic acid | |
| Identifiers | |
3D model (JSmol) | |
| ChEBI | |
| ChemSpider | |
| ECHA InfoCard | 100.007.473 |
| EC Number |
|
| KEGG | |
| MeSH | Methylmalonic+acid |
PubChem CID | |
| UNII | |
CompTox Dashboard (EPA) | |
| |
| |
| Properties | |
| C4H6O4 | |
| Molar mass | 118.088 g/mol |
| Density | 1.455 g/cm−3 |
| Melting point | 134 °C (273 °F; 407 K) |
| Acidity (pKa) | pKa1 = 3,07 [1] pKa2 = 5,76 [1] |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
Methylmalonic acid (MMA) is a chemical compound from the group of dicarboxylic acids. It consists of the basic structure of malonic acid and also carries a methyl group. The salts of methylmalonic acid are called methylmalonates.
