
Alkaptonuria is a rare inherited genetic disease which is caused by a mutation in the HGD gene for the enzyme homogentisate 1,2-dioxygenase ; if a person inherits an abnormal copy from both parents, the body accumulates an intermediate substance called homogentisic acid in the blood and tissues. Homogentisic acid and its oxidized form alkapton are excreted in the urine, giving it an unusually dark color. The accumulating homogentisic acid causes damage to cartilage and heart valves, as well as precipitating as kidney stones and stones in other organs. Symptoms usually develop in people over 30 years old, although the dark discoloration of the urine is present from birth.

Cardiofaciocutaneous (CFC) syndrome is an extremely rare genetic disorder, and is one of the RASopathies. It was first described in 1986.

Menkes disease (MNK), also known as Menkes syndrome, is an X-linked recessive disorder caused by mutations in genes coding for the copper-transport protein ATP7A, leading to copper deficiency. Characteristic findings include kinky hair, growth failure, and nervous system deterioration. Like all X-linked recessive conditions, Menkes disease is more common in males than in females. The disorder was first described by John Hans Menkes in 1962.

Simpson–Golabi–Behmel syndrome (SGBS) is a rare inherited congenital disorder that can cause craniofacial, skeletal, vascular, cardiac, and renal abnormalities. There is a high prevalence of cancer associated in those with SGBS which includes wilms tumors, neuroblastoma, tumors of the adrenal gland, liver, lungs and abdominal organs. The syndrome is inherited in an X-linked recessive manner. Females that possess one copy of the mutation are considered to be carriers of the syndrome but may still express varying degrees of the phenotype, suffering mild to severe malady. Males experience a higher likelihood of fetal death.
Ectrodactyly–ectodermal dysplasia–cleft syndrome, or EEC, and also referred to as EEC syndrome and split hand–split foot–ectodermal dysplasia–cleft syndrome is a rare form of ectodermal dysplasia, an autosomal dominant disorder inherited as a genetic trait. EEC is characterized by the triad of ectrodactyly, ectodermal dysplasia, and facial clefts. Other features noted in association with EEC include vesicoureteral reflux, recurrent urinary tract infections, obstruction of the nasolacrimal duct, decreased pigmentation of the hair and skin, missing or abnormal teeth, enamel hypoplasia, absent punctae in the lower eyelids, photophobia, occasional cognitive impairment and kidney anomalies, and conductive hearing loss.

Laron syndrome (LS), also known as growth hormone insensitivity or growth hormone receptor deficiency (GHRD), is an autosomal recessive disorder characterized by a lack of insulin-like growth factor 1 production in response to growth hormone. It is usually caused by inherited growth hormone receptor (GHR) mutations.
Hay–Wells syndrome is one of at least 150 known types of ectodermal dysplasia. These disorders affect tissues that arise from the ectodermal germ layer, such as skin, hair, and nails.
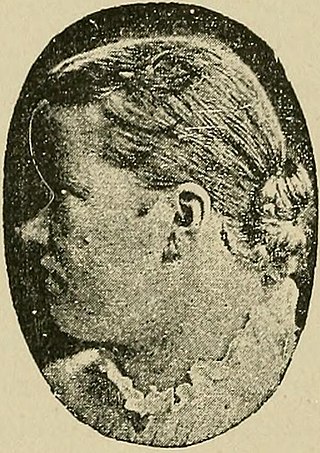
Muenke syndrome, also known as FGFR3-related craniosynostosis, is a human specific condition characterized by the premature closure of certain bones of the skull during development, which affects the shape of the head and face. First described by Maximilian Muenke, the syndrome occurs in about 1 in 30,000 newborns. This condition accounts for an estimated 8 percent of all cases of craniosynostosis.

Uncombable hair syndrome (UHS) is a rare structural anomaly of the hair with a variable degree of effect. It is characterized by hair that is silvery, dry, frizzy, wiry, and impossible to comb. It was first reported in the early 20th century. It typically becomes apparent between the ages of 3 months and 12 years. UHS has several names, including pili trianguli et canaliculi (Latin), cheveux incoiffables (French), and "spun-glass hair". This disorder is believed to be autosomal recessive in most instances, but there are a few documented cases where multiple family members display the trait in an autosomal dominant fashion. Based on the current scientific studies related to the disorder, the three genes that have been causally linked to UHS are PADI3, TGM3, and TCHH. These genes encode proteins important for hair shaft formation. Clinical symptoms of the disorder arise between 3 months and 12 years of age. The quantity of hair on the head does not change, but hair starts to grow more slowly and becomes increasingly "uncombable". To be clinically apparent, 50% of all scalp hair shafts must be affected by UHS. This syndrome only affects the hair shaft of the scalp and does not influence hair growth in terms of quantity, textural feel, or appearance on the rest of the body.

Microvillus inclusion disease, previously known as Davidson's disease, congenital microvillus atrophy and, less specifically, microvillus atrophy, is a rare genetic disorder of the small intestine that is inherited in an autosomal recessive pattern.

Helicase SKI2W is an enzyme that in humans is encoded by the SKIV2L gene. This enzyme is a human homologue of yeast SKI2, which may be involved in antiviral activity by blocking translation of poly(A) deficient mRNAs. The SKIV2L gene is located in the class III region of the major histocompatibility complex.
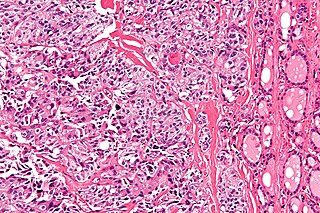
Medullary thyroid cancer is a form of thyroid carcinoma which originates from the parafollicular cells, which produce the hormone calcitonin. Medullary tumors are the third most common of all thyroid cancers and together make up about 3% of all thyroid cancer cases. MTC was first characterized in 1959.
Keutel syndrome (KS) is a rare autosomal recessive genetic disorder characterized by abnormal diffuse cartilage calcification, hypoplasia of the mid-face, peripheral pulmonary stenosis, hearing loss, short distal phalanges (tips) of the fingers and mild mental retardation. Individuals with KS often present with peripheral pulmonary stenosis, brachytelephalangism, sloping forehead, midface hypoplasia, and receding chin. It is associated with abnormalities in the gene coding for matrix gla protein, MGP. Being an autosomal recessive disorder, it may be inherited from two unaffected, abnormal MGP-carrying parents. Thus, people who inherit two affected MGP alleles will likely inherit KS.

Zymogen Granule Protein 16 is a protein that is encoded by the ZG16 gene. Other common names include hZG16, FLJ43571, FLJ92276, secretory lectin ZG16, jacalin-like lectin domain containing, JCLN, JCLN1, MGC183567, MGC34820, ZG16A, zymogen granule membrane protein 16, zymogen granule protein 16 homolog, and zymogen granule protein. The gene is located on Chromosome 16: 29,778,256-29,782,973. The gene obtains one transcript and 128 orthologues.
Trichorrhexis invaginata is a distinctive hair shaft abnormality that may occur sporadically, either in normal hair or with other hair shaft abnormalities, or regularly as a marker for Netherton syndrome. The primary defect appears to be abnormal keratinization of the hair shaft in the keratogenous zone, allowing for intussusception of the fully keratinized and hard distal shaft into the incompletely keratinized and soft proximal portion of the shaft.
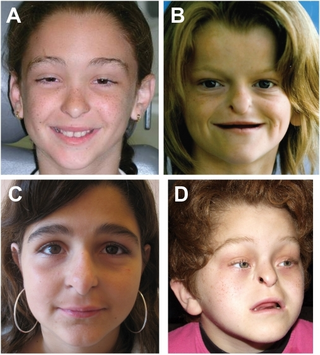
Johanson–Blizzard syndrome (JBS) is a rare, sometimes fatal autosomal recessive multisystem congenital disorder featuring abnormal development of the pancreas, nose and scalp, with intellectual disability, hearing loss and growth failure. It is sometimes described as a form of ectodermal dysplasia.
Congenital tufting enteropathy is an inherited disorder of the small intestine that presents with intractable diarrhea in young children.

Tricho–dento–osseous syndrome (TDO) is a rare, systemic, autosomal dominant genetic disorder that causes defects in hair, teeth, and bones respectively. This disease is present at birth. TDO has been shown to occur in areas of close geographic proximity and within families; most recent documented cases are in Virginia, Tennessee, and North Carolina. The cause of this disease is a mutation in the DLX3 gene, which controls hair follicle differentiation and induction of bone formation. All patients with TDO have two co-existing conditions called enamel hypoplasia and taurodontism in which the abnormal growth patterns of the teeth result in severe external and internal defects. The hair defects are characterized as being rough, course, with profuse shedding. Hair is curly and kinky at infancy but later straightens. Dental defects are characterized by dark-yellow/brownish colored teeth, thin and/or possibly pitted enamel, that is malformed. The teeth can also look normal in color, but also have a physical impression of extreme fragility and thinness in appearance. Additionally, severe underbites where the top and bottom teeth fail to correctly align may be present; it is common for the affected individual to have a larger, more pronounced lower jaw and longer bones. The physical deformities that TDO causes become more noticeable with age, and emotional support for the family as well as the affected individual is frequently recommended. Adequate treatment for TDO is a team based approach, mostly involving physical therapists, dentists, and oromaxillofacial surgeons. Genetic counseling is also recommended.

The HFE H63D is a single-nucleotide polymorphism in the HFE gene, which results in the substitution of a histidine for an aspartic acid at amino acid position 63 of the HFE protein (p.His63Asp). HFE participates in the regulation of iron absorption.

TTC37 is a protein which in humans is encoded by gene TTC37, located on chromosome 5.


