
The Golgi apparatus, also known as the Golgi complex, Golgi body, or simply the Golgi, is an organelle found in most eukaryotic cells. Part of the endomembrane system in the cytoplasm, it packages proteins into membrane-bound vesicles inside the cell before the vesicles are sent to their destination. It resides at the intersection of the secretory, lysosomal, and endocytic pathways. It is of particular importance in processing proteins for secretion, containing a set of glycosylation enzymes that attach various sugar monomers to proteins as the proteins move through the apparatus.

Elastin is a protein encoded by the ELN gene in humans. Elastin is a key component in the extracellular matrix of gnathostomes. It is highly elastic and present in connective tissue of the body to resume its shape after stretching or contracting. Elastin helps skin return to its original position whence poked or pinched. Elastin is also in important load-bearing tissue of vertebrates and used in places where storage of mechanical energy is required.

Proteoglycans are proteins that are heavily glycosylated. The basic proteoglycan unit consists of a "core protein" with one or more covalently attached glycosaminoglycan (GAG) chain(s). The point of attachment is a serine (Ser) residue to which the glycosaminoglycan is joined through a tetrasaccharide bridge. The Ser residue is generally in the sequence -Ser-Gly-X-Gly-, although not every protein with this sequence has an attached glycosaminoglycan. The chains are long, linear carbohydrate polymers that are negatively charged under physiological conditions due to the occurrence of sulfate and uronic acid groups. Proteoglycans occur in connective tissue.

Menkes disease (MNK), also known as Menkes syndrome, is an X-linked recessive disorder caused by mutations in genes coding for the copper-transport protein ATP7A, leading to copper deficiency. Characteristic findings include kinky hair, growth failure, and nervous system deterioration. Like all X-linked recessive conditions, Menkes disease is more common in males than in females. The disorder was first described by John Hans Menkes in 1962.
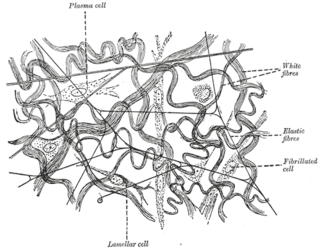
Elastic fibers are an essential component of the extracellular matrix composed of bundles of proteins (elastin) which are produced by a number of different cell types including fibroblasts, endothelial, smooth muscle, and airway epithelial cells. These fibers are able to stretch many times their length, and snap back to their original length when relaxed without loss of energy. Elastic fibers include elastin, elaunin and oxytalan.

Chédiak–Higashi syndrome (CHS) is a rare autosomal recessive disorder that arises from a mutation of a lysosomal trafficking regulator protein, which leads to a decrease in phagocytosis. The decrease in phagocytosis results in recurrent pyogenic infections, albinism, and peripheral neuropathy.

Cutis laxa or pachydermatocele is a group of rare connective tissue disorders in which the skin becomes inelastic and hangs loosely in folds.

SCARF syndrome is a rare syndrome characterized by skeletal abnormalities, cutis laxa, craniostenosis, ambiguous genitalia, psychomotor retardation, and facial abnormalities. These characteristics are what make up the acronym SCARF. It shares some features with Lenz-Majewski hyperostotic dwarfism. It is a very rare disease with an incidence rate of approximately one in a million newborns. It has been clinically described in two males who were maternal cousins, as well as a 3-month-old female. Babies affected by this syndrome tend to have very loose skin, giving them an elderly facial appearance. Possible complications include dyspnea, abdominal hernia, heart disorders, joint disorders, and dislocations of multiple joints. It is believed that this disease's inheritance is X-linked recessive.

Fibrillin-1 is a protein that in humans is encoded by the FBN1 gene, located on chromosome 15. It is a large, extracellular matrix glycoprotein that serves as a structural component of 10–12 nm calcium-binding microfibrils. These microfibrils provide force bearing structural support in elastic and nonelastic connective tissue throughout the body. Mutations altering the protein can result in a variety of phenotypic effects differing widely in their severity, including fetal death, developmental problems, Marfan syndrome or in some cases Weill-Marchesani syndrome.

Fibulin-5 is a protein that in humans is encoded by the FBLN5 gene.

V-type proton ATPase 116 kDa subunit a isoform 2, also known as V-ATPase 116 kDa isoform a2, is an enzyme that in humans is encoded by the ATP6V0A2 gene.
Neuromuscular junction disease is a medical condition where the normal conduction through the neuromuscular junction fails to function correctly.

Gerodermia osteodysplastica (GO) is a rare autosomal recessive connective tissue disorder included in the spectrum of cutis laxa syndromes.

Urbach–Wiethe disease is a very rare recessive genetic disorder, with approximately 400 reported cases since its discovery. It was first officially reported in 1929 by Erich Urbach and Camillo Wiethe, although cases may be recognized dating back as early as 1908.
Anetoderma is a benign but uncommon disorder that causes localized areas of flaccid or herniated sac-like skin due to a focal reduction of dermal elastic tissue. Anetoderma is subclassified as primary anetoderma, secondary anetoderma, iatrogenic anetoderma of prematurity, congenital anetoderma, familial anetoderma, and drug-induced anetoderma.
Cranio-lenticulo-sutural dysplasia is a neonatal/infancy disease caused by a disorder in the 14th chromosome. It is an autosomal recessive disorder, meaning that both recessive genes must be inherited from each parent in order for the disease to manifest itself. The disease causes a significant dilation of the endoplasmic reticulum in fibroblasts of the host with CLSD. Due to the distension of the endoplasmic reticulum, export of proteins from the cell is disrupted.
Inherited disorders of trafficking (IDT) are a family of disorders that involve vesicular delivery of proteins.
De Barsy syndrome is a rare autosomal recessive genetic disorder. Symptoms include cutis laxa as well as other eye, musculoskeletal, and neurological abnormalities. It is usually progressive, manifesting side effects that can include clouded corneas, cataracts, short stature, dystonia, or progeria.
Beare–Stevenson cutis gyrata syndrome is a rare genetic disorder characterized by craniosynostosis and a specific skin abnormality, called cutis gyrata, characterized by a furrowed and wrinkled appearance ; thick, dark, velvety areas of skin are sometimes found on the hands and feet and in the groin.
MEDNIK syndrome(OMIM#609313), also known as "syndrome de Kamouraska", is a genetic disorder that is caused by mutations to the AP1S1 gene. Transmission of the disease is believed to be autosomal recessive. Symptoms of the syndrome are intellectual disability, enteropathy, deafness, neuropathy, ichthyosis, and keratoderma (MEDNIK). People with MEDNIK syndrome often have a high forehead, upslanting palpebral fissures, a depressed nasal bridge, low-set ears, growth retardation, and brain atrophy apparent upon imaging. The disorder was discovered by Patrick Cossette and his research team from the Université de Montréal. MEDNIK syndrome was initially reported in a few French-Canadian families near Quebec who all shared common ancestors.
