
Elias James Corey is an American organic chemist. In 1990, he won the Nobel Prize in Chemistry "for his development of the theory and methodology of organic synthesis", specifically retrosynthetic analysis. Regarded by many as one of the greatest living chemists, he has developed numerous synthetic reagents, methodologies and total syntheses and has advanced the science of organic synthesis considerably.
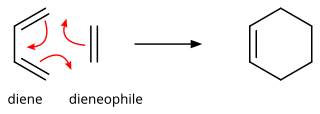
In organic chemistry, the Diels–Alder reaction is a chemical reaction between a conjugated diene and a substituted alkene, commonly termed the dienophile, to form a substituted cyclohexene derivative. It is the prototypical example of a pericyclic reaction with a concerted mechanism. More specifically, it is classified as a thermally-allowed [4+2] cycloaddition with Woodward–Hoffmann symbol [π4s + π2s]. It was first described by Otto Diels and Kurt Alder in 1928. For the discovery of this reaction, they were awarded the Nobel Prize in Chemistry in 1950. Through the simultaneous construction of two new carbon–carbon bonds, the Diels–Alder reaction provides a reliable way to form six-membered rings with good control over the regio- and stereochemical outcomes. Consequently, it has served as a powerful and widely applied tool for the introduction of chemical complexity in the synthesis of natural products and new materials. The underlying concept has also been applied to π-systems involving heteroatoms, such as carbonyls and imines, which furnish the corresponding heterocycles; this variant is known as the hetero-Diels–Alder reaction. The reaction has also been generalized to other ring sizes, although none of these generalizations have matched the formation of six-membered rings in terms of scope or versatility. Because of the negative values of ΔH° and ΔS° for a typical Diels–Alder reaction, the microscopic reverse of a Diels–Alder reaction becomes favorable at high temperatures, although this is of synthetic importance for only a limited range of Diels-Alder adducts, generally with some special structural features; this reverse reaction is known as the retro-Diels–Alder reaction.
In organic chemistry, an electrocyclic reaction is a type of pericyclic rearrangement where the net result is one pi bond being converted into one sigma bond or vice versa. These reactions are usually categorized by the following criteria:
A cascade reaction, also known as a domino reaction or tandem reaction, is a chemical process that comprises at least two consecutive reactions such that each subsequent reaction occurs only in virtue of the chemical functionality formed in the previous step. In cascade reactions, isolation of intermediates is not required, as each reaction composing the sequence occurs spontaneously. In the strictest definition of the term, the reaction conditions do not change among the consecutive steps of a cascade and no new reagents are added after the initial step. By contrast, one-pot procedures similarly allow at least two reactions to be carried out consecutively without any isolation of intermediates, but do not preclude the addition of new reagents or the change of conditions after the first reaction. Thus, any cascade reaction is also a one-pot procedure, while the reverse does not hold true. Although often composed solely of intramolecular transformations, cascade reactions can also occur intermolecularly, in which case they also fall under the category of multicomponent reactions.
A dendralene is a discrete acyclic cross-conjugated polyene. The simplest dendralene is buta-1,3-diene (1) or [2]dendralene followed by [3]dendralene (2), [4]dendralene (3) and [5]dendralene (4) and so forth. [2]dendralene (butadiene) is the only one not cross-conjugated.

The Nicolaou Taxol total synthesis, published by K. C. Nicolaou and his group in 1994 concerns the total synthesis of taxol. Taxol is an important drug in the treatment of cancer but also expensive because the compound is harvested from a scarce resource, namely the pacific yew.

The Petasis reaction is the multi-component reaction of an amine, a carbonyl, and a vinyl- or aryl-boronic acid to form substituted amines.

Paclitaxel total synthesis in organic chemistry is a major ongoing research effort in the total synthesis of paclitaxel (Taxol). This diterpenoid is an important drug in the treatment of cancer but, also expensive because the compound is harvested from a scarce resource, namely the Pacific yew. Not only is the synthetic reproduction of the compound itself of great commercial and scientific importance, but it also opens the way to paclitaxel derivatives not found in nature but with greater potential.

Epothilones are a class of potential cancer drugs. Like taxanes, they prevent cancer cells from dividing by interfering with tubulin, but in early trials, epothilones have better efficacy and milder adverse effects than taxanes.
Wender Taxol total synthesis in organic chemistry describes a Taxol total synthesis by the group of Paul Wender at Stanford University published in 1997. This synthesis has much in common with the Holton Taxol total synthesis in that it is a linear synthesis starting from a naturally occurring compound with ring construction in the order A,B,C,D. The Wender effort is shorter by approximately 10 steps.

Thiostrepton is a natural cyclic oligopeptide antibiotic of the thiopeptide class, derived from several strains of streptomycetes, such as Streptomyces azureus and Streptomyces laurentii. Thiostrepton is a natural product of the ribosomally synthesized and post-translationally modified peptide (RiPP) class.

Pancratistatin (PST) is a natural compound initially extracted from spider lily, a Hawaiian native plant of the family Amaryllidaceae (AMD).

Carpanone is a naturally occurring lignan-type natural product most widely known for the remarkably complex way nature prepares it, and the similarly remarkable success that an early chemistry group, that of Orville L. Chapman, had at mimicking nature's pathway. Carpanone is an organic compound first isolated from the carpano trees of Bougainville Island by Brophy and coworkers, trees from which the natural product derives its name. The hexacyclic lignan is one of a class of related diastereomers isolated from carpano bark as mixtures of equal proportion of the "handedness" of its components, and is notable in its stereochemical complexity, because it contains five contiguous stereogenic centers. The route by which this complex structure is achieved through biosynthesis involves a series of reactions that, almost instantly, take a molecule with little three-dimensionality to the complex final structure. Notably, Brophy and coworkers isolated the simpler carpacin, a phenylpropanoid with a 9-carbon framework, recognized its substructure as being dimerized within the complex carpanone structure, and proposed a hypothesis of how carpacin was converted to carpanone in plant cells:

Betaenone B, like other betaenones, is a secondary metabolite isolated from the fungus Pleospora betae, a plant pathogen. Its phytotoxic properties have been shown to cause sugar beet leaf spots, which is characterized by black, pycnidia containing, concentric circles eventually leading to necrosis of the leaf tissue. Of the seven phytotoxins isolated in fungal leaf spots from sugar beet, betaenone B showed the least amount of phytotoxicity showing only 8% inhibition of growth while betaenone A and C showed 73% and 89% growth inhibition, respectively. Betaenone B is therefore not considered toxic to the plant, but will produce leaf spots when present in high concentrations (0.33 μg/μL). While the mechanism of action of betaenone B has yet to be elucidated, betaenone C has been shown to inhibit RNA and protein synthesis. Most of the major work on betaenone B, including the initial structure elucidation of betaenone A, B and C as well as the partial elucidation mechanism of biosynthesis, was presented in three short papers published between 1983–88. The compounds were found to inhibit a variety of protein kinases signifying a possible role in cancer treatment.

Strychnine total synthesis in chemistry describes the total synthesis of the complex biomolecule strychnine. The first reported method by the group of Robert Burns Woodward in 1954 is considered a classic in this research field.
The retro-Diels–Alder reaction is the reverse of the Diels–Alder (DA) reaction, a [4+2] cycloelimination. It involves the formation of a diene and dienophile from a cyclohexene. It can be accomplished spontaneously with heat, or with acid or base mediation.

Torreyanic acid is a dimeric quinone first isolated and by Lee et al. in 1996 from an endophyte, Pestalotiopsis microspora. This endophyte is likely the cause of the decline of Florida torreya, an endangered species that is related to the taxol-producing Taxus brevifolia. The natural product was found to be cytotoxic against 25 different human cancer cell lines with an average IC50 value of 9.4 µg/mL, ranging from 3.5 (NEC) to 45 (A549) µg/mL. Torreyanic acid was found to be 5-10 times more potent in cell lines sensitive to protein kinase C (PKC) agonists, 12-o-tetradecanoyl phorbol-13-acetate (TPA), and was shown to cause cell death via apoptosis. Torreyanic acid also promoted G1 arrest of G0 synchronized cells at 1-5 µg/mL levels, depending on the cell line. It has been proposed that the eukaryotic translation initiation factor EIF-4a is a potential biochemical target for the natural compound.
Metal-catalyzed C–H borylation reactions are transition metal catalyzed organic reactions that produce an organoboron compound through functionalization of aliphatic and aromatic C–H bonds and are therefore useful reactions for carbon–hydrogen bond activation. Metal-catalyzed C–H borylation reactions utilize transition metals to directly convert a C–H bond into a C–B bond. This route can be advantageous compared to traditional borylation reactions by making use of cheap and abundant hydrocarbon starting material, limiting prefunctionalized organic compounds, reducing toxic byproducts, and streamlining the synthesis of biologically important molecules. Boronic acids, and boronic esters are common boryl groups incorporated into organic molecules through borylation reactions. Boronic acids are trivalent boron-containing organic compounds that possess one alkyl substituent and two hydroxyl groups. Similarly, boronic esters possess one alkyl substituent and two ester groups. Boronic acids and esters are classified depending on the type of carbon group (R) directly bonded to boron, for example alkyl-, alkenyl-, alkynyl-, and aryl-boronic esters. The most common type of starting materials that incorporate boronic esters into organic compounds for transition metal catalyzed borylation reactions have the general formula (RO)2B-B(OR)2. For example, bis(pinacolato)diboron (B2Pin2), and bis(catecholato)diborane (B2Cat2) are common boron sources of this general formula.
Biomimetic synthesis is an area of organic chemical synthesis that is specifically biologically inspired. The term encompasses both the testing of a "biogenetic hypothesis" through execution of a series of reactions designed to parallel the proposed biosynthesis, as well as programs of study where a synthetic reaction or reactions aimed at a desired synthetic goal are designed to mimic one or more known enzymic transformations of an established biosynthetic pathway. The earliest generally cited example of a biomimetic synthesis is Sir Robert Robinson's organic synthesis of the alkaloid tropinone.
The Danheiser benzannulation is a chemical reaction used in organic chemistry to generate highly substituted phenols in a single step. It is named after Rick L. Danheiser who developed the reaction.




![Initial electrocyclizations in Nicolaou endiandric acid C synthesis, to provide diol 17 shown in the main scheme. This key intermediate is then ready for elaboration into the olefin which can undergo the further 6p[4+2] (Diels-Alder) cycloaddition reaction shown in the main scheme, to provide the title natural product. ElectrocyclizationInEndrianicAcidSynth.svg](http://upload.wikimedia.org/wikipedia/commons/thumb/0/0a/ElectrocyclizationInEndrianicAcidSynth.svg/350px-ElectrocyclizationInEndrianicAcidSynth.svg.png)