Related Research Articles

Alpha-mannosidosis is a lysosomal storage disorder, first described by Swedish physician Okerman in 1967. In humans it is known to be caused by an autosomal recessive genetic mutation in the gene MAN2B1, located on chromosome 19, affecting the production of the enzyme alpha-D-mannosidase, resulting in its deficiency. Consequently, if both parents are carriers, there will be a 25% chance with each pregnancy that the defective gene from both parents will be inherited, and the child will develop the disease. There is a two in three chance that unaffected siblings will be carriers. In livestock alpha-mannosidosis is caused by chronic poisoning with swainsonine from locoweed.

Calnexin (CNX) is a 67kDa integral protein of the endoplasmic reticulum (ER). It consists of a large N-terminal calcium-binding lumenal domain, a single transmembrane helix and a short, acidic cytoplasmic tail. In humans, calnexin is encoded by the gene CANX.

Mannosidosis is a deficiency in mannosidase, an enzyme. There are two types: alpha-mannosidosis and beta-mannosidosis. Both disorders are related to the lysosome and have similar presentation; the former is caused by defective lysosomal α-mannosidase and the latter by defective lysosomal β-mannosidase. In both cases, the defect causes accumulation of oligosaccharides rich in mannose in the neural tissue and organ tissue. Both alpha- and beta-mannosidosis are known to result from autosomal recessive genetic mutations.
α-Mannosidase is an enzyme involved in the cleavage of the α form of mannose. Its systematic name is α-D-mannoside mannohydrolase.

Endoplasmic-reticulum-associated protein degradation (ERAD) designates a cellular pathway which targets misfolded proteins of the endoplasmic reticulum for ubiquitination and subsequent degradation by a protein-degrading complex, called the proteasome.

Endoplasmic reticulum mannosyl-oligosaccharide 1,2-alpha-mannosidase is an enzyme that in humans is encoded by the MAN1B1 gene.

Alpha-mannosidase 2 is an enzyme that in humans is encoded by the MAN2A1 gene.

Glycoprotein endo-alpha-1,2-mannosidase is an enzyme that in humans is encoded by the MANEA gene.

Epididymis-specific alpha-mannosidase is an enzyme that in humans is encoded by the MAN2B2 gene.

Mannosyl-oligosaccharide 1,2-alpha-mannosidase IA is an enzyme that in humans is encoded by the MAN1A1 gene.
β-Mannosidase is an enzyme with systematic name β-D-mannoside mannohydrolase, which is in humans encoded by the MANBA gene. This enzyme catalyses the following chemical reaction

Beta-mannosidosis, also called lysosomal beta-mannosidase deficiency, is a disorder of oligosaccharide metabolism caused by decreased activity of the enzyme beta-mannosidase. This enzyme is coded for by the gene MANBA, located at 4q22-25. Beta-mannosidosis is inherited in an autosomal recessive manner. Affected individuals appear normal at birth, and can have a variable clinical presentation. Infantile onset forms show severe neurodegeneration, while some children have intellectual disability. Hearing loss and angiokeratomas are common features of the disease.

Mannosyl-oligosaccharide 1,2-alpha-mannosidase IB is an enzyme that in humans is encoded by the MAN1A2 gene.
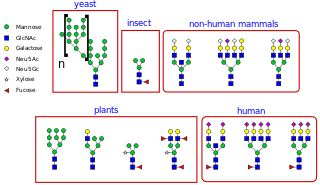
N-linked glycosylation is the attachment of an oligosaccharide, a carbohydrate consisting of several sugar molecules, sometimes also referred to as glycan, to a nitrogen atom, in a process called N-glycosylation, studied in biochemistry. The resulting protein is called an N-linked glycan, or simply an N-glycan.
Congenital dyserythropoietic anemia type II, or hereditary erythroblastic multinuclearity with positive acidified serum lysis test (HEMPAS) is a rare genetic anemia in humans characterized by hereditary erythroblastic multinuclearity with positive acidified serum lysis test.
Mannosyl-oligosaccharide 1,2-α-mannosidase (EC 3.2.1.113, mannosidase 1A, mannosidase 1B, 1,2-α-mannosidase, exo-α-1,2-mannanase, mannose-9 processing α-mannosidase, glycoprotein processing mannosidase I, mannosidase I, Man9-mannosidase, ManI, 1,2-α-mannosyl-oligosaccharide α-D-mannohydrolase) is an enzyme with systematic name 2-α-mannosyl-oligosaccharide α-D-mannohydrolase. It catalyses the hydrolysis of the terminal (1→2)-linked α-D-mannose residues in the oligo-mannose oligosaccharide Man9(GlcNAc)2.
Mannosyl-oligosaccharide 1,3-1,6-α-mannosidase, also known as Golgi α-mannosidase II, is an enzyme with systematic name (1→3)-(1→6)-mannosyl-oligosaccharide α-D-mannohydrolase. It catalyses the hydrolysis of the terminal (1→3)- and (1→6)-linked α-D-mannose residues in the mannosyl-oligosaccharide Man5(GlcNAc)3.
Glycoprotein endo-α-1,2-mannosidase (EC 3.2.1.130, glucosylmannosidase, endo-α-D-mannosidase, endo-α-mannosidase, endomannosidase, glucosyl mannosidase) is an enzyme with systematic name glycoprotein glucosylmannohydrolase. It catalyses the hydrolysis of the terminal α-D-glucosyl-(1,3)-D-mannosyl unit from the GlcMan9(GlcNAc)2 oligosaccharide component of the glycoprotein produced in the Golgi membrane.
Mannosylglycoprotein endo-beta-mannosidase is an enzyme. This enzyme catalyses the following chemical reaction:

Kifunensine is an alkaloid originally isolated from Kitasatosporia kifunense, an actinobacterium. It is a neutral, stable compound.
References
- ↑ Mannosidases at the U.S. National Library of Medicine Medical Subject Headings (MeSH)