This article includes a list of general references, but it lacks sufficient corresponding inline citations .(February 2013) |

An orchidometer (or orchiometer) is a medical instrument used to measure the volume of the testicles.
This article includes a list of general references, but it lacks sufficient corresponding inline citations .(February 2013) |
An orchidometer (or orchiometer) is a medical instrument used to measure the volume of the testicles.

The orchidometer was introduced in 1966 by Swiss pediatric endocrinologist Andrea Prader of the University of Zurich. [1] It consists of a string of twelve numbered wooden or plastic beads of increasing size from about 1 to 25 millilitres. Doctors sometimes informally refer to them as "Prader's balls", "the medical worry beads", or the "endocrine rosary". [2]
The beads are compared with the testicles of the patient, and the volume is read off the bead which matches most closely in size. Prepubertal sizes are 1–3 ml, pubertal sizes are considered 4 ml and up and adult sizes are 15-25 ml. [2]
The orchidometer can be used to accurately determine size of testes. Discrepancy of testicular size with other parameters of maturation can be an important clue to various diseases. Small testes can indicate either primary or secondary hypogonadism. Testicular size can help distinguish between different types of precocious puberty. Since testicular growth is typically the first physical sign of true puberty, one of the most common uses is as confirmation that puberty is beginning in a boy with delayed puberty. Large testes (macroorchidism) can be a clue to one of the most common causes of inherited generalised learning disability, fragile X syndrome.
Stephen Shalet, a leading endocrinologist who works for the Christie Hospital in Manchester, is reported to have told The Observer : "Every endocrinologist should have an orchidometer. It's his stethoscope."[ citation needed ]
Orchidometers are also commonly used to measure testicular volume of rams.
Numerous clinical scales and measurement systems exist to define genitals as normal male or female, or "abnormal", including the Prader scale, Quigley scale, and the satirical Phall-O-Meter.[ citation needed ]

A testicle or testis is the gonad in all male bilaterians, including humans, and is homologous to the ovary in females. Its primary functions are the production of sperm and the secretion of androgens, primarily testosterone.

Testicular torsion occurs when the spermatic cord twists, cutting off the blood supply to the testicle. The most common symptom in children is sudden, severe testicular pain. The testicle may be higher than usual in the scrotum and vomiting may occur. In newborns, pain is often absent and instead the scrotum may become discolored or the testicle may disappear from its usual place.

Testicular cancer is cancer that develops in the testicles, a part of the male reproductive system. Symptoms may include a lump in the testicle or swelling or pain in the scrotum. Treatment may result in infertility.

Cryptorchidism, also known as undescended testis, is the failure of one or both testes to descend into the scrotum. The word is from Ancient Greek κρυπτός (kryptos) 'hidden' and ὄρχις (orchis) 'testicle'. It is the most common birth defect of the male genital tract. About 3% of full-term and 30% of premature infant boys are born with at least one undescended testis. However, about 80% of cryptorchid testes descend by the first year of life, making the true incidence of cryptorchidism around 1% overall. Cryptorchidism may develop after infancy, sometimes as late as young adulthood, but that is exceptional.
Delayed puberty is when a person lacks or has incomplete development of specific sexual characteristics past the usual age of onset of puberty. The person may have no physical or hormonal signs that puberty has begun. In the United States, girls are considered to have delayed puberty if they lack breast development by age 13 or have not started menstruating by age 15. Boys are considered to have delayed puberty if they lack enlargement of the testicles by age 14. Delayed puberty affects about 2% of adolescents.
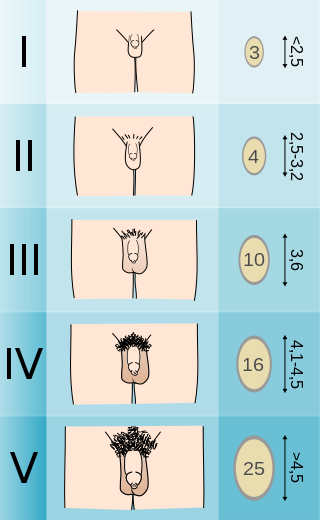
The Tanner scale is a scale of physical development as pre-pubescent children transition into adolescence, and then adulthood. The scale defines physical measurements of development based on external primary and secondary sex characteristics, such as the size of the breasts, length of the penis, volume of the testes, and growth of pubic hair. This scale was first quantified in 1969 by James Tanner, a British pediatrician, after a two-decade-long study following the physical changes in girls undergoing puberty.
Hypogonadism means diminished functional activity of the gonads—the testicles or the ovaries—that may result in diminished production of sex hormones. Low androgen levels are referred to as hypoandrogenism and low estrogen as hypoestrogenism. These are responsible for the observed signs and symptoms in both males and females.

Spermatocele is a fluid-filled cyst that develops in the epididymis. The fluid is usually a clear or milky white color and may contain sperm. Spermatoceles are typically filled with spermatozoa and they can vary in size from several millimeters to many centimeters. Small spermatoceles are relatively common, occurring in an estimated 30 percent of males. They are generally not painful. However, some people may experience discomfort such as a dull pain in the scrotum from larger spermatoceles. They are not cancerous, nor do they cause an increased risk of testicular cancer. Additionally, unlike varicoceles, they do not reduce fertility.

Testicular atrophy is a medical condition in which one or both testicles diminish in size and may be accompanied by reduced testicular function. Testicular atrophy is not related to the temporary shrinkage of the surrounding scrotum, which might occur in response to cold temperature.
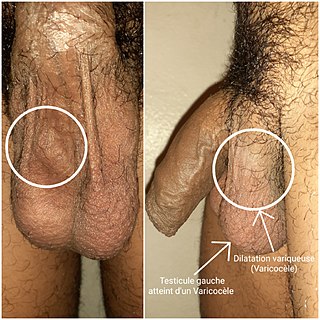
A varicocele is, in a man, an abnormal enlargement of the pampiniform venous plexus in the scrotum; in a woman, it is an abnormal painful swelling to the embryologically identical pampiniform venous plexus; it is more commonly called pelvic compression syndrome. In the male varicocele, this plexus of veins drains blood from the testicles back to the heart. The vessels originate in the abdomen and course down through the inguinal canal as part of the spermatic cord on their way to the testis. Varicoceles occur in around 15% to 20% of all men. The incidence of varicocele increase with age.
Macroorchidism is a disorder found in males, specifically in children, where a subject has abnormally large testes. The condition is commonly inherited in connection with fragile X syndrome (FXS), which is also the second most common genetic cause of intellectual disability. The condition is also a rare sign of McCune–Albright syndrome. The opposite of macroorchidism is called microorchidism, which is the condition of abnormally small testes.
Male infertility refers to a sexually mature male's inability to impregnate a fertile female. In humans, it accounts for 40–50% of infertility. It affects approximately 7% of all men. Male infertility is commonly due to deficiencies in the semen, and semen quality is used as a surrogate measure of male fecundity. More recently, advance sperm analyses that examine intracellular sperm components are being developed.
Gonadal dysgenesis is classified as any congenital developmental disorder of the reproductive system characterized by a progressive loss of primordial germ cells on the developing gonads of an embryo. One type of gonadal dysgenesis is the development of functionless, fibrous tissue, termed streak gonads, instead of reproductive tissue. Streak gonads are a form of aplasia, resulting in hormonal failure that manifests as sexual infantism and infertility, with no initiation of puberty and secondary sex characteristics.
Testicular sperm extraction (TESE) is a surgical procedure in which a small portion of tissue is removed from the testicle and any viable sperm cells from that tissue are extracted for use in further procedures, most commonly intracytoplasmic sperm injection (ICSI) as part of in vitro fertilisation (IVF). TESE is often recommended to patients who cannot produce sperm by ejaculation due to azoospermia.

Epididymal cyst is a harmless sac in the testicles filled with fluid. The most frequent clinical presentation occurs when a routine physical examination yields an unexpected finding, which is then confirmed by scrotal ultrasonography. Although the exact cause of epididymal cysts is unknown, it is likely a congenital anomaly associated with hormonal imbalances during the embryonic stage of development.
Puberty is the process of physical changes through which a child's body matures into an adult body capable of sexual reproduction. It is initiated by hormonal signals from the brain to the gonads: the ovaries in a female, the testicles in a male. In response to the signals, the gonads produce hormones that stimulate libido and the growth, function, and transformation of the brain, bones, muscle, blood, skin, hair, breasts, and sex organs. Physical growth—height and weight—accelerates in the first half of puberty and is completed when an adult body has been developed. Before puberty, the external sex organs, known as primary sexual characteristics, are sex characteristics that distinguish males and females. Puberty leads to sexual dimorphism through the development of the secondary sex characteristics, which further distinguish the sexes.

Orchiectomy is a surgical procedure in which one or both testicles are removed. The surgery can be performed for various reasons:
Hypergonadotropic hypogonadism (HH), also known as primary or peripheral/gonadal hypogonadism or primary gonadal failure, is a condition which is characterized by hypogonadism which is due to an impaired response of the gonads to the gonadotropins, follicle-stimulating hormone (FSH) and luteinizing hormone (LH), and in turn a lack of sex steroid production. As compensation and the lack of negative feedback, gonadotropin levels are elevated. Individuals with HH have an intact and functioning hypothalamus and pituitary glands so they are still able to produce FSH and LH. HH may present as either congenital or acquired, but the majority of cases are of the former nature. HH can be treated with hormone replacement therapy.

Leydig cell hypoplasia (LCH), also known as Leydig cell agenesis, is a rare autosomal recessive genetic and endocrine syndrome affecting an estimated 1 in 1,000,000 individuals with XY chromosomes. It is characterized by an inability of the body to respond to luteinizing hormone (LH), a gonadotropin which is normally responsible for signaling Leydig cells of the testicles to produce testosterone and other androgen sex hormones. The condition manifests itself as pseudohermaphroditism, hypergonadotropic hypogonadism, reduced or absent puberty, and infertility.
Male genital examination is a physical examination of the genital in males to detect ailments and to assess sexual development, and is normally a component of an annual physical examination. The examination includes checking the penis, scrotum, and urethral meatus. A comprehensive assessment of the male genitals assesses the pubic hair based on Sexual Maturity Rating and the size of the testicles and penis. The exam can also be conducted to verify a person's age and biological sex. The genitourinary system can also be assessed as part of the male genital examination. During a genital examination, the doctor can detect any of the following: structural abnormalities, urethral opening abnormalities, problems related to not being circumcised, lumps, tumors, redness, excoriation, edema, lesions, swelling, cancer, hair-related issues, and many others. In some instances where a physical examination of the male genitals is not sufficient to diagnose an individual, then an internal genital examination using imaging or ultrasounds will be needed for further evaluation.
| Human rights and legal issues | ||
|---|---|---|
| Healthcare and biology | ||
| Society and culture | ||
| History and events | ||
| Rights by country | ||
| See also | ||